Измерение - удельная активность
Cтраница 2
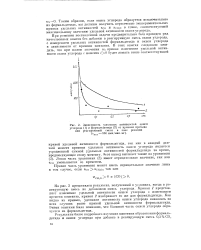 |
Зависимость удельных активностей окиси. [16] |
При решении поставленной задачи предварительно был проведен ряд качественных опытов без добавки в реагирующую смесь окиси углерода, с измерением удельных активностей формальдегида и окиси углерода в зависимости от времени контакта. [17]
Погрешность измерения составляет 35 % номинального значения поддиапазона. Время измерения минимальной удельной активности не превышает 20 мин. [18]
Общая точность результата зависит от точности определения удельной активности, которая должна быть выполнена особенно тщательно. Препараты для измерения удельной активности соединения и исходного раствора должны быть приготовлены одинаковым способом, иметь одинаковую толщину, а все измерения активности выполнены в одинаковых условиях. [19]
Все препараты для измерения активности готовят и измеряют в одинаковых условиях, выпариванием определенного объема раствора в стандартных чашечках, а в случаях возможного образования летучих соединений или тогда, когда объемы, подлежащие упариванию, очень велики, применяют метод осаждения с носителями. Промежуток времени между измерениями удельной активности образцов должен быть мал по сравнению с периодом полураспада изотопа, иначе следует вводить поправку на распад. [20]
Подобным методом были произведены абсолютные измерения удельной активности этилового и метилового спиртов и получены хорошо воспроизводимые результаты. Таким методом можно производить измерения удельной активности любого ji - активного газа или жидкости, имеющей невысокую температуру кипения, при условии, что они не вступают в реакцию с деталями детектора и не растворяются в смазке вакуумных кранов системы. [21]
Бензоаты представляют собой белые кристаллические вещества с характерными температурами плавления, которые сравнительно легко выделяются из сложной органической смеси. Из них нетрудно приготовить хорошие образцы для измерения удельной активности. [22]
Количество экстрагированного металла остается одинаковым и для пробы, и для эталона. В методе изотопного разбавления удается существенно понизить предел определения, так как отпадает необходимость IB измерении удельной активности и для расчета нужны лишь - активности исследуемого и эталонного экстрактов. Не нужно также определять массу выделенного компонента, что позволяет использовать метод изотопного разбавления в случае нанограммовых количеств. [23]
Сопоставление данных, полученных в различных исследованиях, проведенное Бударом [101], показывает согласие величин удельной активности катализаторов разного генезиса по начальным скоростям реакций, соответственно, для процессов гидрирования, дегидрирования и изомеризации. Полторак [195] полагает, что в области размеров частиц, превышающей 50А, где проводились в основном измерения удельной активности нанесенных катализаторов, ее различий вообще нельзя было обнаружить в виду малых изменений среднего координационного числа поверхностных атомов. Однако такое утверждение, хотя и относится только к структурному аспекту постоянства удельной активности, как раз и является доводом в пользу справедливости правила Борескова и расчетов теории абсолютных скоростей. [24]
При однократной экстракции для экстракции возможно большего количества интересующего элемента обычно используют избыток реагента. Однако субстехиометрическое количество комплексообразующего реагента, например дитизона или купферрона, с успехом применяют в методе изотопного разбавления для экстракции постоянного и предельно малого количества таких элементов, как Zn, Hg, Си и Fe, при измерениях удельной активности. [25]
Образовавшийся осадок отделяют центрифугированием, промывают 5 % - ным раствором нитрата калия, подкисленного уксусной кислотой. Промытый осадок растворяют соляной кислотой в центрифужной пробирке при нагревании, доводят объем раствора в пробирке до определенного деления ( 5 - 10 мл), отбирают 1 мл полученного раствора в стеклянную чашку для измерения активности, сушат под лампой и измеряют активность остатка на том же счетчике, на котором производилось измерение удельной активности нитрата кобальта. Из полученных данных рассчитывают количество калия. [26]
Далее сорбент извлекается из капсулы и промывается дистиллированной водой. На этом заканчивается стадия концентрирования, которая ввиду высокой селективности сорбции включает также групповое разделение элементов и в значительной степени - радиохимическую очистку. Десорбцией или растворением сорбента стронций или цезий переводятся в раствор, производится определение содержания стабильного изотопа и измерение удельной активности раствора. [27]
Вырезают этот участок бумаги и измеряют скорость счета на торцовом счетчике. Оставшиеся части хроматограммы разделяют на участки 1 5 см я измеряют для каждого из них скорость счета. Для определения относительной активности компонентов необходимо просуммировать скорость счета отрезков бумаги, превышающих фон в два раза, и отнести скорость счета каждого из них к сумме скоростей счета всей хроматограммы. Измерение удельной активности препарата проводят с помощью общепринятой методики. [28]
В оставшемся первоначальном растворе определяют аналитически весовое содержание элемента и удельную активность радиоактивного вещества. Активность находят следующим путем. Из первоначального раствора отбирают 1 мл в мерную колбу и доводят объем до 100 мл. Препарат для измерения удельной активности должен быть приготовлен таким же способом, как проба из насыщенного раствора. [29]
Метод изотопного разбавления [1] позволяет количественно разделять и определять сходные элементы или их соединения в смеси друг с другом. Этот метод не требует количественного выделения определяемого компонента. Расчет содержания исходного компонента ведется по результатам измерения удельной активности при разбавлении в ходе анализа. [30]
Страницы: 1 2 3