Учет - корреляционная энергия
Cтраница 1
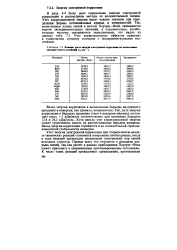 |
Влияние учета энергия электронной корреляции на вычисленные значении частот колебаний со, ( см 1. [1] |
Учет корреляционной энергии имеет важное значение при определении формы потенциальных кривых и поверхностей. Так, вычисленные длины связей в методе Хартри-Фока оказываются короче экспериментальных значений, а следовательно, колебательные частоты оказываются завышенными, что видно из данных табл. 7.3. Учет корреляционных эффектов приводит к существенно лучшему согласию с экспериментальными значениями. [2]
Другой путь учета корреляционной энергии состоит в том, что вообще отказываются от понятия одноэлектронного состояния. Вместо этого в качестве основы для описания выбираются двух-электронные волновые функции, или геминали. [3]
 |
Расчеты структуры и энергии циклобутадиена в основных триплетном и синглетном состояниях3. [4] |
Первая величина получена в расчете ССП, вторая - с учетом корреляционной энергии в приближении связанных электронных пар. [5]
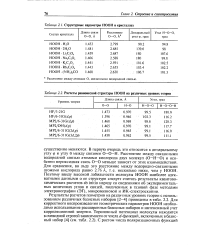 |
Структурные параметры НООН в кристаллах.| Расчеты равновесной структуры НООН на различных уровнях теории. [6] |
Результаты расчетов геометрии на различных уровнях теории с использованием различных базисных наборов [2-4] приведены в табл. 2.2. Для корректного воспроизведения геометрических параметров НООН необходимо использование расширенных базисных наборов и интенсивный учет корреляционной энергии. [7]
 |
Схема нахождения наиболее устойчивой геометрии катиона FHC CH. [8] |
Наиболее строгие последние расчеты с базисом STO-6 - 31G предсказывают, что винильный катион более устойчив в мости-ковой ( 16) форме, а этильный в классической ( Па) форме, дальнейшее улучшение расчета путем учета корреляционной энергии ( см. раздел 4.4) не изменяет результата. [9]
Все полуэмпирические методы приводят к выводу о большей стабильности мо-стиковой формы. Учет корреляционной энергии разными методами приводит к определенному выводу о большей устойчивости неклассической мости-ковой формы этильного катиона [39, 40], причем открытая форма переходит в мостиковую без барьера. Структуры, которые разные методы предсказывают для C2Hs, согласуются между собой. В мостиковой форме связь С-С несколько удлинена по сравнению со свободным этиленом ( на 0 006 - 0 007 нм), а длина связи С - Н ОСт составляет 0 129 - 0 133 нм. В классической структуре связь С-С удлинена несколько больше ( на 0 007 - 0 012 нм), но все еще гораздо короче, чем в этане. Длины связей С - Н различаются между собой, но незначительно. Как неэмпирические ( STO - 3G - 6 - 31 - G), так и полуэмпирические ( MINDO / 3) расчеты определенно предсказывают, что единственной стабильной ациклической структурой является 2-пропильный катион. Структуры с мостиковым атомом Н нестабильны, а 1-пропильный катион без ак-тивационного барьера переходит в структуру, представляющую собой циклопропан, протонирозанный по вершине. [10]
Возможно конструирование ИДР таким образом, что тепловой эффект равен разности энергий диссоциации одной и той же связи в исследуемом и реперном соединениях. В этом случае взаимная компенсация погрешностей неучтенной Екар особенно эффективна, что позволяет использовать небольшие базисные наборы и невысокий уровень учета корреляционной энергии. Этот подход дает возможность рассчитывать органические соединения существенно больших размеров и получать надежные значения важнейшей термодинамической величины-энергии диссоциации химической связи. [11]
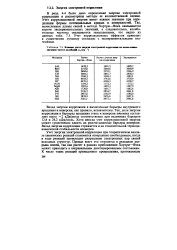 |
Влияние учета энергия электронной корреляции на вычисленные значении частот колебаний со, ( см 1. [12] |
Вклад энергии корреляции в вычисленные барьеры внутреннего вращения и инверсии, как правило, незначителен. Так, доля энергии корреляции в барьерах вращения этана и инверсии акшиака составляет всего - 2 кДж / моль соответственно при величинах барьеров 12 4 и 24 2 кДж / моль. Хотя иногда учет корреляционной энергии может существенно влиять на рассчитываемые барьеры инверсии. Вклад энергии корреляции отражается и на относительной термодинамической стабильности изомеров. [13]
Главным недостатком неэмпирических расчетов по методу Хартри-Фока - Рутаана является неучет электронной корреляции. Учет электронной корреляции методами конфигурационного взаимодействия, теории возмущений или функционала плотности дает лучшие результаты, но, как отмечалось выше, требует значительных временных и машинных ресурсов. Разработан ряд подходов к учету корреляционной энергии. Применение этих подходов позволяет учесть большую часть ( более 95 %) корреляционной энергии. [14]
Вплоть до 0 12 нм подход ( 0, 90) приводит к монотонному понижению энергии, причем в этой области подходы с меньшими значениями р и большими значениями а Становятся выгоднее и система без всякого барьера должна переходить в конечную структуру. Отметим следующие обстоятельства, представляющие значительный интерес. Во-первых, даже для несогласованных подходов учет конфигурационного взаимодействия может приводить не только к количественному, но - и к качественному изменению результатов. ССП приводит к повышению энергии системы, тогда как при учете корреляционной энергии знак эффекта меняется. Во-вторых, результаты расчета полуэмпирическим методом CNDO в параметризации Фишера - - Колльмара качественно согласуются с результатами неэмтаирическото расчета, хотя количественные различия велики. [15]
Страницы: 1 2