Полный учет - конфигурационное взаимодействие
Cтраница 1
Полный учет конфигурационного взаимодействия при использовании ограниченного базисного набора дает точное решение уравнения Шредингера, возможное для этого базисного набора. Следовательно, степень воспроизведения этого результата в рамках применяемой модели позволяет судить о ее точности, хотя при сопоставлении базисных наборов чрезвычайно важное значение имеет погрешность, связанная с тем, как обрывают разложение. [1]
Однако столь полный учет конфигурационного взаимодействия на заданном конечном базисе неэкономичен, так как требует больших затрат машинного времени. [2]
 |
Типичные числа Вейля. [3] |
Вообще говоря, полный учет конфигурационного взаимодействия при использовании базисных наборов приемлемых размеров представляет собой задачу, невыполнимую в вычислительном отношении, поскольку число членов в разложении ( 7.2 1) для волновой функции чрезвычайно велико. [4]
С другой стороны, при полном учете конфигурационного взаимодействия, при полном учете всех тех конфигурационных функций состояния, которые могут быть построены на том или ином наборе молекулярных орбиталей, становится неважным, как эти орбитали преобразуются друг через друга, т.е. в каком конкретно виде они заданы. По этой причине в многоконфигурационных функциях стараются сначала ввести такие молекулярные орбитали, с которыми ряд метода конфигурационного взаимодействия сходится наиболее быстро, далее из полученного таким образом набора молекулярных орбиталей отбирают лишь некоторую часть наиболее существенных орбиталей, и на этом более узком наборе пользуются методом полного конфигурационного взаимодействия, в рамках которого вполне очевиден переход к локализованным орбиталям того или иного типа. [5]
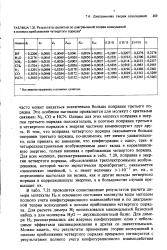 |
Результаты расчетов по диаграммной теории возмущений в полном приближении четвертого порядка. [6] |
В табл. 7.21 проводится сопоставление результатов расчета ди-мера молекулы Н2 и основного состояния молекулы воды методом полного учета конфигурационного взаимодействия и по диаграммной теории возмущений в полном приближении четвертого порядка. [7]
В заключение надо отметить, что расчет спектра с использованием самосогласованных орбиталей и с более или менее полным учетом конфигурационного взаимодействия во всех случаях приводит к хорошему соответствию с экспериментом. [8]
Этот результат имеет общий характер: если исходить из заданного базисного набора, то молекулярно-орбитальный подход с полным учетом конфигурационного взаимодействия приводит к таким же результатам, как и метод валентных связей с полным учетом конфигурационного взаимодействия. [9]
Этот результат имеет общий характер: если исходить из заданного базисного набора, то молекулярно-орбитальный подход с полным учетом конфигурационного взаимодействия приводит к таким же результатам, как и метод валентных связей с полным учетом конфигурационного взаимодействия. [10]
За исключением случая очень малых систем, неполнота базисного набора почти всегда является важнейшим источником погрешностей в молекулярных расчетах, а также в большинстве расчетов атомов, где используется алгебраическое приближение. Никакое расширение конфигурационного взаимодействия или суммирование членов высших порядков ряда возмущений не может компенсировать погрешности, связанной с неполнотой базисного набора. Результаты, полученные при полном учете конфигурационного взаимодействия в пространстве прямых произведений N-то ранга, генерируемого заданным базисным набором, могут по мере достижения полноты базисного набора лишь приближаться к результату полного учета конфигурационного взаимодействия. Выбор базисного набора в исследованиях атомных и молекулярных систем оказывает определяющее влияние на точность расчета. [11]
 |
Энергия взаимодействия двух молекул Н2. [12] |
Принципиальная трудность связана с тем, что дисперсионную энергию можно описать лишь при помощи методов, в которых учитывается энергия корреляции. Таким образом, в принципе невозможно вычисление дисперсионной энергии в рамках метода Хартри - Фока. При использовании вариационного подхода удается получить удовлетворительные результаты в рамках неэмпирического метода ССП в сочетании с полным учетом конфигурационного взаимодействия; еще одна возможность заключается в проведении расчетов с гамильтонианом, в который включена межэлектронная координата. При менее точных расчетах достаточно ограничиться учетом двукратно возбужденных конфигураций, однако и в этом случае для молекул, состоящих из нескольких атомов, объем вычислений очень велик. [13]
 |
Энергия взаимодействия двух молекул Нг. [14] |
Принципиальная трудность связана с тем, что дисперсионную энергию можно описать лишь при помощи методов, в которых учитывается энергия корреляции. Таким образом, в принципе невозможно вычисление дисперсионной энергии в рамках метода Хартри-Фока. При использовании вариационного подхода удается получить удовлетворительные результаты в рамках неэмпирического метода ССП в сочетании с полным учетом конфигурационного взаимодействия; еще одна возможность заключается в проведении расчетов с гамильтонианом, в который включена межэлектронная координата. При менее точных расчетах достаточно ограничиться учетом двукратно возбужденных конфигураций, однако и в этом случае для молекул, состоящих из нескольких атомов, объем вычислений очень велик. [15]
Страницы: 1 2