Уэстхеймер
Cтраница 1
Уэстхеймер [9] рассмотрел случай многоосиовиых кислот, В которых проточи структурно це эквивалентны. [1]
Перрен и Уэстхеймер [254] показали, что этот эффект обусловлен активацией иона ртути, происходящей, аналогично активации ионов водорода, при удалении воды из сольватной оболочки. Термодинамическая активность растворителя ( воды) уменьшается при добавлении электролита. [2]
Кирквуду и Уэстхеймеру [14] принадлежит наиболее удачная попытка в разработке такой модели, в которой некоторая часть пространства была бы занята молекулой, а некоторая его часть - растворителем; такая модель позволяет отдельно вычислить член, отвечающий электростатической части работы. Конечно, допущение, согласно которому молекула имеет точную эллипсоидальную форму ( причем происходит локализация центров полярности и диссоциации внутри эллипсоида), а также допущение, что диэлектрическая проницаемость равна 2, являются произвольными в этой теории; однако такие допущения дают возможность одинаково подходить к рассмотрению различных по длине и толщине молекул. Все попытки расчета влияния поля в количественном отношении вызывают сомнение, однако имеется путь, который позволяет качественно судить о существовании эффекта поля. Необходимо выбрать такую систему, чтобы эффект поля конкурировал с внутренней передачей полярного влияния по механизму сопряжения и чтобы было известно, что эффект поля имеет противоположное направление; тогда при достаточно большой величине эффекта поля последний может перекрыть эффект сопряжения. Можно привести два примера, в которых внутренний полярный эффект имеет одно направление, а эффект поля - противоположное. Такие опыты были проведены Роберт-сом и сотрудниками. Известно, что передаваемый внутримолекулярно полярный эффект сопряжения в бензольном кольце проявляется сильнее в более удаленном / гяра-положении, чем в более близком лштга-положении. [3]
В том же году Уэстхеймер и Майер [85] применили сходную по идее схему расчета скорости рацемизации оптически деятельных замещенных дифенилов. Именно эти расчеты показали, что стерическое напряжение в молекуле в первую очередь приводит к искажению валентных углов по сравнению с углами в ненапряженной молекуле. Так, например, как указал Коулсон [74], согласно расчетам его и Лонгет-Хиггинса ( 1951 г.), основной деформацией, сводящей к минимуму взаимное отталкивание атомов водорода в дифениле, является поворот колец вокруг центральной связи СС. [4]
Такой вывод соответствует данным Уэстхеймера, который показал, что при ароматическом меркурировании медленной стадией будет отщепление протона; в данном случае реакция происходит в обратном направлении. [5]
Как отмечалось в классических работах Уэстхеймера [339], любой тип вандерваальсова сжатия в молекуле приводит к ослаблению его путем деформации молекулы, так как искажения углов и торсионные деформации приводят к намного меньшему увеличению энергии молекулы, чем вандерваальсовы взаимодействия. [6]
Почти одновременно с Хиллом и независимо от него Уэстхеймер и Майер [36] разработали математическую теорию для аналогичного принципа нахождения минимума энергии и показали, как этот метод мог бы применяться к вычислению энергии активации рацемизации оптически деятельных произведших бифенила. Их метод и расчеты рассмотрены здесь подробно. [7]
Он разработал приемлемую вандерваальсову функцию ( 46J для взаимодействия метпльных групп и распространил методы, ранее разработанные Уэстхеймером и Майором. Он вычислил, что избыток пространственной энергии в случае i / мс-нзомера составляет 1 85 ккал / моль; экспериментальная величина, полученная Кистяков-ским из теплот гидрогенизации [47], составляет 1 29 ккал / моль. Совпадение результатов достаточно хорошее. [8]
Авторы определили спектрофотометрически равновесие ионизации протонированной воды, являвшейся лигандом окта-эдрического комплекса трехвалентного железа гемов метагемоглобина крови и дали теоретическое объяснение процессу, исходя из диэлектрической теории Кирквуда и Уэстхеймера. Известны гемоглобины, отличающиеся только одним пептидным звеном в определенном положении определенной полипептидной цепи. [9]
Высота барьера заторможенного вращения дается приближенно экспериментальной энергией активации рацемизации, определяемой из температурного коэффициента реакции при помощи уравнения Аррениуса. Уэстхеймер и другие рассчитали высоты барьеров на основании данных о потенциалах отталкивания между ограничивающими вращение атомами и группами и силовых постоянных связей, растяжение и изгиб которых может уменьшить выигрыш энергии межатомного притяжения в плоской конфигурации. Результаты расчета были позднее подтверждены другими группами исследователей. [10]
Исследователи из Университетского колледжа ( Лондон) улучшили [34] свои более ранние вычисления 127 ] энергии активации реакции замещения. В 1946 - 1947 гг. Уэстхеймер и Майер показали ту решающую роль, которую играет изгибание в пространственных факторах ( см. стр. Рацемизация оптически деятельных бифенилов); в этом новом исследовании [34] принимается во внимание изгиб валентных связей. По необходимости эти вычисления очень сложны, так как сведение к минимуму энергии переходного состояния ( см. Математический метод) требует согласования многих длин связей и углов и отбора предпочтительных вращательных кон-формаций для замещающих групп. Далее, эти вычисления неизбежно несколько неопределенны, так как они основаны не только на вандер-ваальсовых потенциалах, но и на таких труднодоступных количественных данных, как силовые константы для изгиба и растяжения шолусвязей и как энергия взаимодействия растворителя с ионными участниками различного рода. [11]
Теория электронных смещений не является единственно возможным подходом к проблеме связи строения и реакционной способности. Например, теория электрического поля, первоначально предложенная Бьеррумом [64] и усовершенствованная Кирквудом и Уэстхеймером [65], объясняет соотношение первой и второй константы ионизации двухосновных кислот тем, что отрицательный заряд СОСГ-группы в однозарядном анионе увеличивает работу удаления протона от оставшейся группы ОООН. [12]
Далее, поскольку скорость декарбо-ксилирования соединения I в присутствии брома или йода идентична скорости присоединения галоида, было сделано заключение, что отщепление углекислоты приводит непосредственно к образованию енолыюй формы соединения, которая затем мгновенно реагирует с галоидом. Хотя эти факты находятся в согласии с распадом диполярной формы исходной: кислоты, как сообщал Педерсен, Уэстхеймер [2] показал, что дскарбоксилироиание р-кетокислот в отсутствие катализатора не зависит от диэлектрической постоянной растворителя и что поэтому в стадию, которая определяет скорость реакции, не входит сильно полярное промежуточное соединение. [13]
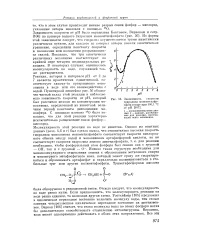 |
Зависимость скорости гидролиза мопометилфосфата в воде при 100 1 С от рН. 1 -экспериментальные данные. 2 -данные, рассчитанные для реакции, протекающей через моноанион. [14] |
Отсюда следует, что молекулярность по воде равна нулю. Если предположить, что молекулярность реакции по воде равна единице, то возможна другая схема. Уэстхеймер [105] предложил в циклическое переходное состояние включить молекулу воды, тем самым заменив четырехзвенное циклическое переходное состояние на шестизвен-ное. Верной [106] считает, что атака молекулы воды по атому фосфора могла бы дополнительно способствовать отщеплению метоксигруппы. Вероятно, вода может одновременно действовать в обоих этих направлениях. [15]
Страницы: 1 2