Гидрирование - этиленовая связь
Cтраница 2
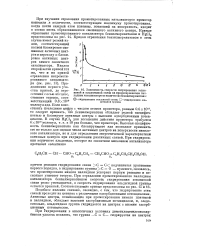 |
Зависимость скорости гидрирования этиленовой и альдегидной связи на промотированном палладием катализаторе от количества бензилмеркаптана. [16] |
С С подчиняется уравнению первого порядка, а гидрирование группы С 0 - нулевого, оказалось, что промотирование никеля палладием ускоряет первую реакцию и несколько угнетает вторую. При отравлении промотированного палладием катализатора бензилмеркаптаном скорость гидрирования этиленовой связи резко уменьшается, а скорость гидрирования альдегидной группы остается прежней. [17]
В самом деле, как видно из цифр табл. 5, MoS2 резко разграничивает углеводороды по скорости их гидрирования лишь в границах целого класса соединений, определяемого типом двойной связи. При этом оказывается справедливой общая для Ni и MoS2 закономерность: скорость гидрирования этиленовой связи больше скорости гидрирования связи конденсированной ароматики, а последней - больше скорости гидрирования связей бензольного кольца. [18]
Энергия образования непредельных соединений, содержащих гидррксильный или эфирный кислород при ненасыщенном углеродном атоме, больше, чем энергия образования изомерных им соединений, в которых кислород находится при насыщенном углеродном атоме. Вывод о большей энергии образования этих соединений может быть сделан на основании сравнения теплот гидрирования этиленовой связи в углеводородах и кислородсодержащих соединениях указанного типа ( табл. 27; ср. [19]
Энергия образования непредельных соединений, содержащих гидроксильный или эфирный кислород при ненасыщенном углеродном атоме, больше, чем энергия образования изомерных им соединений, в которых кислород находится при насыщенном углеродном атоме. Вывод о большей энергии образования этих соединений может быть сделан на основании сравнения теплот гидрирования этиленовой связи в углеводородах и кислородсодержащих соединениях указанного типа ( табл. 24, ср. В табл. 24 приведена теплота гидрирования аллилового спирта, в котором кислородный атом стоит при насыщенном углеродном атоме; теплота гидрирования его близка к теплоте гидрирования пропилена. [20]
Транс-цкклооктен реагирует энергично с фени-лазидом, что свидетельствует о сильном напряжении в молекуле. Теплота гидрирования двойной связи составляет 32 24 0 21 ккал / моль [42], что является одной из наиболее высоких тсплот гидрирования этиленовой связи, измеренных до сих пор. Теплота изомеризации транс-циклооктена в цис-соединении составляет 9 24 ккал / моль и также является необыкновенно высокой. Для циклодецена она составляет только 3 3 ккал / моль. Эти данные свидетельствуют о присутствии большего напряжения в молекуле транс-циклооктена. [21]
Солективное гидрирование двойной углерод-углеродной связи с сохранением карбонильной группы легко осуществить для кетонов, функциональная группа которых менее реакционноспособна, чем в альдегидах. Катализаторами могут служить платина, никель, медь и другие металлические, но не оксидные контакты. Условия процесса существенно не отличаются от применяемых при гидрировании олефинов, но при выборе условий следует учитывать возможное побочное восстановление кетонной группы. В случае ненасыщенных альдегидов гидрирование только этиленовой связи предггавляет собой более сложную задачу. Для этого необходимы возмэжно более мягкие условия и катализаторы, мало активные в отношении гидрирования карбонильных групп. [22]
Селективное гидрирование двойной углерод-углеродной связи с сохранением карбонильной группы легко осуществить для кетонов, функциональная группа которых менее реакционно-способна, чем в альдегидах. Катализаторами могут служить платиновые, никелевые, медные и другие металлические, но не оксидные контакты. Условия процесса существенно не отличаются от применяемых при гидрировании олефинов, но при выборе условий следует учитывать возможное побочное восстановление кетонной группы. В случае ненасыщенных альдегидов гидрирование только этиленовой связи представляет собой более сложную задачу. Для этого необходимы возможно более мягкие условия и катализаторы, малоактивные в отношении-гидрирования карбонильных групп. [23]
Селективное гидрирование двойной углерод-углеродной связи с сохранением карбонильной группы легко осуществить для кетонов функциональная группа которых менее реакционноспособна, чем в альдегидах. Катализаторами могут служить платина, никель, медь и другие металлические, но не окисные контакты. Условия процесса не отличаются существенно от применяемых при гидрировании олефинов, но при их выборе следует учитывать возможное побочное восстановление кетонной группы. В случае ненасыщенных альдегидов гидрирование только этиленовой связи представляет собой более сложную задачу. Для этого необходимы возможно более мягкие условия и катализаторы, малоактивные в отношении гидрирования карбонильных групп. [24]
Селективное гидрирование двойной углерод-углеродной связи с сохранением карбонильной группы легко осуществить для кето-нов, функциональная группа которых менее реакцйонноспособна, чем в альдегидах. Катализаторами могут служить платина, никель, медь и другие металлические, но не окисные контакты. Условия процесса не отличаются существенно от применяемых при гидрировании олефинов, но при их выборе следует учитывать возможное побочное восстановление кетонной группы. В случае ненасыщенных альдегидов гидрирование только этиленовой связи представляет собой более сложную задачу. Для этого необходимы возможно более мягкие условия и катализаторы, мало активные в отношении гидрирования карбонильных групп. [25]
Обычные лабораторные опыты с 20 - 30 г вещества или с еще меньшими количествами удобнее всего проводятся с помощью одной из описанных выше ( стр. Во многих случаях может быть необходимо проследить за присоединением водорода. Лишь изредка можно быть вполне уверенным в полной безвредности избыточного водорода; с хорошими катализаторами обыкновенно через некоторое время гидрируется также и ароматическое кольцо. При небольших загрузках расход водорода определяется лучше всего по объему, при больших - манометрически, о чем все необходимое было сказано выше. Гидрирование этиленовых связей под давлением, как это делается в большем масштабе при гидрогенизации жиров, в лаборатории излишне. Простой пример может дать представление о том, как в настоящее время производятся подобные опыты. [26]
Определение теплот активации для реакций гидрирования цнклогсксена, циклогексадиенов и бензола на платине в условиях, при которых кажущиеся теплоты активации не включают теплоты адсорбции этих соединений, приводит к выводу [367], что в присутствии катализатора резонансная энергия бензола исчезает. Теплота активации в реакции гидрирования бензола ( 7 4 ккал / мол) весьма близка к эндотермическому эффекту ( 5 6 - 7 4 ккал / г-мол), который является минимальной энергией активации, требуемой для гидрирования бензола в циклический диен. Установлено [167], что при адсорбции на катализаторе происходит разрыв л-связей. Вследствие этого перегруппировка, сопровождающая хеыосорбцию бензола, приводит к дополнительному увеличению энергии активации ( около 34 ккал / г-мол) по сравнению с наблюдаемой при хемосорбции олефина; этой потерей энергии и объясняется трудность гидрирования бензола. Аналогичные рассуждения могут быть приведены для других ароматических систем. Таким образом можно объяснить то обстоятельство, что многие катализаторы, активные в реакциях гидрирования этиленовых связей, оказываются неактивными при гидрировании ароматических ядер. [27]
Страницы: 1 2