Свободный гидроксиламин
Cтраница 3
Низшие гидроксиламины обладают типичным аминным запахом и представляют собой жидкости или твердые вещества. Гидроксиламины обладают основными свойствами и образуют соли с минеральными кислотами ( например, гидрохлориды и гидробромиды), можно также получить оксалаты и пикраты гидроксиламинов. Во многих случаях эти соли гораздо более устойчивы, чем свободные гидроксиламины, и гидроксиламины предпочитают хранить в виде солей. Увеличение степени замещения при атоме азота и кислорода понижает основность, как и в случае производных гидразина. [31]
Наличие в молекуле нитрила электроноакцепторных заместителей значительно ускоряет реакцию. Присоединение гидроксиламина ускоряется, например, положительно заряженным атомом азота в а-положении к нитрильной группе. В связи с этим при получении амидоксимов из аминоацетонитрилов целесообразно исходить не из свободного гидроксиламина, а из его гидрохлорида. [32]
В кислых растворах NH2OH находится в протонированной форме [ NH3OH ], которая не может взаимодействовать с кетоном. Поэтому при достаточной кислотности раствора определяющей скорость стадией становится присоединение оставшегося непротонированного NHjOH, за которой следует быстрая стадия катализируемой кислотой дегидратации. Следовательно, в кислом растворе скорость превращения должна быть ниже максимальной из-за низкой концентрации свободного гидроксиламина. [33]
В недавних работах также высказано предположение, что при соблюдении условий, относящихся к форме молекул и правильной ориентации связывающих групп, могут быть допущены значительные вариации в отношении разрываемых при реакции связей и даже самого характера катализируемой реакции. Так, про-теазы ( пептидазы) атакуют не только пептидные связи, но и эфирные связи в сложных эфирах, обладающих конфигурацией, подобной пептидам. Некоторые карбоксильные эстеразы гидроли-зуют не только эфиры карбоновых кислот, но и аналогичные по структуре эфиры фосфорной кислоты; другие энзимы катализируют синтез гидроксамовых кислот из эфиров карбоновых кислот и свободного гидроксиламина. [34]
Это предположение может вызвать некоторое недоумение, поскольку каждый распад промежуточного соединения приводит к образованию продукта и константа скорости первого порядка для распада промежуточного продукта в реакции, которая подчиняется общему первому порядку, должна быть равна константе скорости всей реакции. Однако существенным является то, что абсолютная скорость изменения концентрации промежуточного соединения, накапливающегося в заметной степени, выраженная в моль / л - мин, является очень небольшой по сравнению со скоростью изменения концентраций реагентов и продуктов. Для реакции гидроксиламина стационарное рассмотрение кинетики реакции можно применить при исследовании реакции в условиях низких значений рН, в которых большая часть гидроксиламина протонирует-ся и но происходит заметного накапливания промежуточного продукта; стационарную кинетику, однако, нельзя использовать для анализа экспериментальных данных при высоких значениях рН, когда концентрация свободного гидроксиламина достаточно высока и способна превратить существенную долю карбонильного соединения в промежуточный продукт присоединения. [35]
Наиболее заманчивой реакций для определения карбонильной группы является реакция с использованием гидроксиламина. Использование фенилгидра-зина и других замещенных гидразинов всегда требует отделения твердого производного с последующим анализом либо избытка реагента, либо твердого производного. Последний вариант метода не пригоден для субмикроколичеств, так как он и утомителен и неточен. При добавлении точно измеренного количества органического основания ( 2-диметиламиноэтанола) к раствору гидрохлорида гидроксиламина выделяется известное количество свободного гидроксиламина; после добавления карбонильного соединения и полного оксимирования избыток гидроксиламина титруют хлорной кислотой. Реакция проходит в неводной среде, гидрохлорид гидроксиламина и органическое основание растворяют в пропаноле-2, а титрование проводят раствором хлорной кислоты в метилцеллозольве. [36]
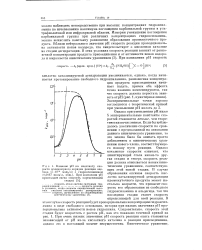
Однако при уменьшении рН ниже 5 экспериментальные константы скоростей становятся меньше, чем теоретически вычисленные. Если бы наблюдалось увеличение скорости по сравнению с предсказанной на основании данного кинетического уравнения, то это можно было бы описать просто добавлением к кинетическому уравнению нового члена, соответствующего новому пути реакции. Однако понижение скорости означает, что лимитирующей стала какая-то другая стадия и теперь скорость реакции должна описываться новым кинетическим уравнением, соответствующим этой новой стадии. В случае образования оксимов скорость кислотно катализируемой дегидратации промежуточного продукта может настолько возрасти, что превысит скорость его образования из свободного гидроксиламина и альдегида, так что последняя стадия станет скорость определяющей для всей реакции. [38]
Метод применим для большинства алифатических и ароматических альдегидов. При исследовании хлорзамещенных алифатических альдегидов - могут получаться повышенные результаты вследствие отщепления хлора. Из кетонов обычно количественно оксимируются только алифатические кетоны, например ацетон, метилэтилкетон, метилгексилкетон, метил-изо-гексилкетон, метилнонилкетон, дигептилкетон, диоктилкетон, динонилкетон, циклопентанон, цибетон, ацетофенон, бензилацетон. Другие кетоны, как, например, изобутирон и бензофенон, реагируют лишь в очень небольшой степени. При анализе подобных карбонильных соединений, характеризующемся обычно возвращением первоначальной окраски оттитрованного раствора, для получения удовлетворительных результатов следует пользоваться методом оксимирования свободным гидроксиламином при нагревании. [39]
Метод применим для большинства алифатических и ароматических альдегидов. При исследовании хлорзамещенных алифатических альдегидов могут получаться повышенные результаты вследствие отщепления хлора. Из кетонов обычно количественно оксимируются только алифатические кетоны, например ацетон, метилэтилкетон, метилгексилкетон, метил-зо-гексилкетон, метилнонилкетон, дигептилкетон, диоктилк он, динонилкетон, циклопентанон, цибетон, ацетофенон, бензилацетон. Другие кетоны, как, например, изобутирон и бензофенон, реагируют лишь в очень небольшой степени. При анализе подобных карбонильных соединений, характеризующемся обычно возвращением первоначальной окраски оттитрованного раствора, для получения удовлетворительных результатов следует пользоваться методом оксимирования свободным гидроксиламином при нагревании. [40]
Поскольку этот фермент использует в качестве восстановителя виологен, можно ожидать, что донором электронов будет служить также ферредок-син. Далее было показано, что восстановленный пиридиннуклеотид не может служить в данном случае донором водорода; следовательно, исключено участие в этом процессе сопряженной системы переноса водорода от пиридиннуклео-тида к ферредоксину. При очистке фермента необходимо присутствие сульфгидрильных соединений, причем фермент чувствителен к цианиду и ацетату фенилртути. Следовательно, в функционировании фермента принимают участие SH-грушш и, возможно, ионы тяжелого металла. Фермент восстанавливает гидроксиламин, но со значительно меньшей скоростью, чем это свойственно общей реакции восстановления нитрита до аммиака. Это говорит о том, что в качестве промежуточного продукта в этой реакции не образуется свободный гидроксиламин. [41]
Страницы: 1 2 3