Грегор
Cтраница 1
Грегор с сотрудниками исследовали линейный полиэтиленглицин на комплексообразованиес медью, потенциометрически титровали его, исследовали вязкость и электрофоретическую подвижность. Они нашли, что этот полимер является высоко полярным веществом. Даже в изоэлектрической точке, которая лежит в интервале рН 2 7 - 2 9, эта полимерная аминокислота все еще легко растворима в воде. Поли - М - этиленглицин обладает слабым сродством к щелочным ионам. Из результатов потенциометрического титрования полиамфолитов в присутствии ионов Си 2 следует, что образуется устойчивый комплекс состава 1: 2 [ ср. &2 12 04), в то время как третья молекула глицина фиксируется весьма слабо. [1]
Грегор, Сандхейм, Хельд и Уаксман [10] определили изменение объема 10 различных катионных форм 10 % ДВБ суль-фокислотой смолы при семи различных значениях относительной влажности, вплоть до насыщения. Их результаты показывают, что увеличение объема матрицы при сорбции воды происходит для различных катионных форм одинаковым образом. Это приблизительно соответствует пространству, которое занимали бы молекулы воды в гипотетическом состоянии плотной упаковки. При добавлении следующих молекул воды парциальный молярный объем возрастает, пока при содержании воды около 10 моль / г-экв не достигается такое состояние, что добавляемая вода занимает практически свой нормальный объем. Это дает основание рассматривать среду между цепями как обычную водную фазу. [2]
Грегор [25] первый четко указал на то, что явление ионного обмена в смолах можно рассматривать как пример доннанов-ского равновесия, причем осмотическое давление внутреннего раствора уравновешивается механическим давлением напряженной полимерной сетки. Основная идея этой концепции, однако, принадлежит Проктору и Вильсону ( 1916 г.), которые использовали ее для объяснения влияния рН на набухание желатины. Незаряженный сшитый гель набухает в подходящем растворителе до равновесного объема, определяемого главным образом отношением объема, приходящегося на одну поперечную связь, к молярному объему растворителя и величиной термодинамического коэффициента взаимодействия между полимером и растворителем; для степени набухания можно вывести термодинамические или статистические формулы ( см., например, [26]), причем свободная энергия смешивания уравновешивается свободной энергией растяжения полимерных цепочек. Эта теория, однако, не нашла применения для ионообменных смол, используемых на практике, во-первых, ввиду того, что степень их сшивки достаточно высока, а во-вторых, потому, что свободная энергия растворения сшитой смолы относительно невелика по сравнению с энергией смешивания растворителя с диссоциированными ионами. Если рассматривать фазу набухшей смолы как раствор, то станет очевидным, что молярная концентрация ионов несравнимо больше, чем концентрация полимера; таким образом, ответственными за понижение химического потенциала растворителя в среде являются главным образом противоионы. Следовательно, вода из внешнего раствора имеет тенденцию диффундировать внутрь до тех пор, пока энергия растяжения матрицы не скомпенсирует противоположный по знаку член в выражении для свободной энергии. [3]
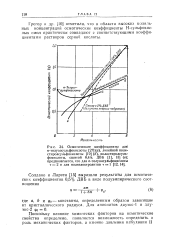
Грегор и др. [10] отметили, что в области высоких моляль-ных концентраций осмотические коэффициенты Н - сульфокис-лых смол практически совпадают с соответствующими коэффициентами растворов серной кислоты. [5]
Грегор, Белл и Маркус [23], например, утверждали, что поскольку большие анионы принято считать негидратированными, то обнаруженный ими эффект уменьшения объема трихлораце-татной смолы по сравнению с ацетатной формой можно хорошо объяснить, если допустить, что первая смола склонна к образованию ионных пар. Даже n - толуолсульфоформа набухала меньше, чем ацетатная, - в согласии с известной ван-дер-ваальсов-ской сорбцией ароматических ионов. [6]
Грегор, Сандхейм, Хельд и Уаксман [10] определили изменение объема 10 различных катионных форм 10 % ДВБ суль-фокислотой смолы при семи различных значениях относительной влажности, вплоть до насыщения. Их результаты показывают, что увеличение объема матрицы при сорбции воды происходит для различных катионных форм одинаковым образом. Это приблизительно соответствует пространству, которое занимали бы молекулы воды в гипотетическом состоянии плотной упаковки. При добавлении следующих молекул воды парциальный молярный объем возрастает, пока при содержании воды около 10 моль / г-экв не достигается такое состояние, что добавляемая вода занимает практически свой нормальный объем. Это дает основание рассматривать среду между цепями как обычную водную фазу. [7]
Грегор [25] первый четко указал на то, что явление ионного обмена в смолах можно рассматривать как пример доннанов-ского равновесия, причем осмотическое давление внутреннего раствора уравновешивается механическим давлением напряженной полимерной сетки. Основная идея этой концепции, однако, принадлежит Проктору и Вильсону ( 1916 г.), которые использовали ее для объяснения влияния рН на набухание желатины. Незаряженный сшитый гель набухает в подходящем растворителе до равновесного объема, определяемого главным образом отношением объема, приходящегося на одну поперечную связь, к молярному объему растворителя и величиной термодинамического коэффициента взаимодействия между полимером и растворителем; для степени набухания можно вывести термодинамические или статистические формулы ( см., например, [26]), причем свободная энергия смешивания уравновешивается свободной энергией растяжения полимерных цепочек. Эта теория, однако, не нашла применения для ионообменных смол, используемых на практике, во-первых, ввиду того, что степень их сшивки достаточно высока, а во-вторых, потому, что свободная энергия растворения сшитой смолы относительно невелика по сравнению с энергией смешивания растворителя с диссоциированными ионами. Если рассматривать фазу набухшей смолы как раствор, то станет очевидным, что молярная концентрация ионов несравнимо больше, чем концентрация полимера; таким образом, ответственными за понижение химического потенциала растворителя в среде являются главным образом противоионы. Следовательно, вода из внешнего раствора имеет тенденцию диффундировать внутрь до тех пор, пока энергия растяжения матрицы не скомпенсирует противоположный по знаку член в выражении для свободной энергии. [8]
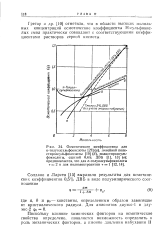
Грегор и др. [10] отметили, что в области высоких моляль-ных концентраций осмотические коэффициенты Н - сульфокис-лых смол практически совпадают с соответствующими коэффициентами растворов серной кислоты. [10]
Грегор, Белл и Маркус [23], например, утверждали, что поскольку большие анионы принято считать негидратированными, то обнаруженный ими эффект уменьшения объема трихлораце-татной смолы по сравнению с ацетатной формой можно хорошо объяснить, если допустить, что первая смола склонна к образованию ионных пар. Даже л-толуолсульфоформа набухала меньше, чем ацетатная, - в согласии с известной ван-дер-ваальсов-ской сорбцией ароматических ионов. [11]
Грегор и Врегман [48] опубликовали кривые титрования для ряда смол известного химического состава, которые определенно показывают, что некоторые смолы содержат два или большее число типов кислотных групп, характеризующихся отчетливыми точками перегиба на кривых нейтрализации. Кривые скорости обмена при различных значениях рН показывают, что сильно кислотные группы нейтрализуются быстрее, чем слабо кислотные группы. [12]
Грегор понимал, что в приведенном выше уравнении не учитывается обычное межионное взаимодействие, которое следует ожидать в очень концентрированных растворах электролитов, и ввел дополнительный член, отражающий влияние этого взаимодействия. Глкжауф критиковал теорию Грегора на том основании, что член ионного взаимодействия значительно превышает осмотический член. Рассматривая ионообменные смолы как концентрированные растворы электролитов и применяя правило Харнэда, Глюкауф оценил величину члена ионного взаимодействия и получил хорошее согласование с экспериментальными данными по избирательности. В обеих концепциях подчеркивается решающее влияние упругих свойств смолы на ионообменное равновесие с тем различием, что Грегор исходит из прямого влияния упругих сил на осмотический член, а Глюкауф - из их косвенного влияния на член ионного взаимодействия. [13]
Грегор подошел к понятию избирательности с точки зрения термодинамики, объяснив ее разницей в давлении набухания и в молярных объемах ионов в смоле. Широкое применение нашла точка зрения Эйхорна, который рассматривал ионообменник как концентрированный раствор электролита, исходя из чего Бойд и Глюкауф успешно применили к ионному обмену учение об активности. [14]
Грегор с сотрудниками ( Брегман) объясняют изменение порядка сорбируемости ионов щелочных металлов на фосфорных и карбоксильных смолах в сравнении с сульфосмолами на обратный изменением порядка поляризуемости. [15]
Страницы: 1 2 3 4