Использование - базисный набор
Cтраница 2
В табл. 6.13 иллюстрируется важная роль высших гармоник для молекулярных систем. Здесь представлены результаты расчетов молекулы моноксида углерода с использованием базисных наборов функций s -, р - и d - симметрии, а также базисных наборов функций s -, p -, d - и f - симметрии [748] и показана зависимость энергий матричного метода Хартри - Фока и корреляционных энергий, полученных диаграммным методом теории возмущений, от геометрии ядер. [17]
С годами накопился большой материал, позволяющий судить о способности определенных базисных наборов обеспечивать заданную точность в рамках заданной модели. Тот факт, что расчеты матричным методом Хартри - Фока с использованием базисных наборов двухэкспонентного качества приводят к хорошим результатам для длин связей, представляет собой эмпирическое наблюдение, основанное на многих расчетах. Можно было бы утверждать, что такие результаты объяснимы компенсацией между поляризационными эффектами и корреляционными эффектами, но это утверждение невозможно строго обосновать теоретически. Однако из этого вовсе не следует, что подобные эмпирические наблюдения бесполезны. Действительно, выбор базисного набора может рассматриваться как часть концепции теоретической модели химии ( см, разд. Если конкретный метод, использующий определенный базисный набор, был применен к целому кругу проблем и при этом были установлены его точность и применимость к решению этих проблем, то этот метод приобретает определенную предсказательную способность. Подобный подход широко распространен в применениях квантово-механических методик к изучению молекул. [18]
В табл. 6.8 приведены результаты применения выражений (6.10.9) - (6.10.13) к атомам некоторых элементов второго периода. Эти результаты получены на основе расчетов [617], выполненных в рамках модели Хартри - Фока с использованием базисных наборов гауссовых функций. [19]
Следует отметить, что вычисляемый вклад в энергию, обусловленный переносом заряда, сильно зависит от качества исходной волновой функции и заметно снижается по мере ее уточнения, например по мере расширения базисного набора. Первые две строки содержат результаты расчета по теории возмущений [9] и по методу МО ЛКАО ССП [10] с использованием минимального базисного набора. В двух последних строках приведены данные расчетов по теории возмущений [11] и по методу МО ЛКАО ССП [5] с привлечением сравнительно широкого базисного набора; результаты последнего находятся в хорошем согласии с экспериментом. [20]
 |
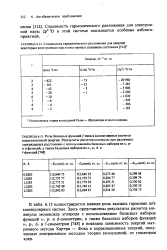
Типичные числа Вейля. [21] |
В табл. 7.1 приведены типичные числа Вейля Dm. Очевидно, что расчет основного состояния десятиэлектронной системы ( например, молекулы воды) с полным учетом конфигурационного-взаимодействия при использовании достаточно большого базисного набора, скажем из 75 базисных функций, остался бы практически нереализуемым даже в том случае, если бы возможности современной вычислительной техники чрезвычайно возросли. [22]
Следует, однако, отметить, что в расчетах неустойчивых частиц в рамках алгебраического приближения существует возможность получения случайных решений. Например, отрицательный ион, который в действительности неустойчив к распаду на нейтральную частицу и свободный электрон, можно ошибочно счесть устойчивым на основании расчета с использованием ограниченного базисного набора. Важнейший источник возможной погрешности при использовании алгебраического приближения заключается в предположении, что решение существует. [23]
Например, равновесная геометрия ядер молекулы непосредственно определяется электронной энергией. Равновесная геометрия, определенная с использованием ограниченного базисного набора при неполном учете электронной корреляции или вовсе без него, часто приводит к довольно точным значениям длин связей. Такое согласие с экспериментальными данными чаще всего оказывается не случайным. Расширение базисного набора в матричном методе Хартри - Фока обычно приводит к несколько меньшим длинам связи по сравнению с экспериментальными значениями. Учет корреляционных эффектов приводит к возрастанию вычисленных длин связей и в результате к лучшему согласию с экспериментальными значениями. Учет корреляционной энергии имеет важное значение также при определении формы потенциальных кривых и поверхностей. Учет корреляционной энергии может заметно влиять на вычисляемые разности энергий, например барьеры внутреннего вращения или потенциалы ионизации. [24]
Метод ЛКАО можно было бы считать точным, если бы для построения истинной молекулярной орбитали использовались все возможные атомные орбитали. Но, учитывая чрезмерную сложность такой задачи, обычно в качестве базисного набора выбирают небольшое число атомных орбиталей, что делает метод ЛКАО приближенным. Простейшее рассмотрение молекулы водорода основано на использовании базисного набора из двух атомных ls - орбиталей. [25]
Подавляющее большинство современных неэмпирических расчетов проводится, если можно так выразиться, с прагматическим подходом: в них не определяются границы погрешностей и точность расчета устанавливается на основе сопоставления с величинами, извлекаемыми из эксперимента. Таким образом, устанавливается качество конкретного базисного набора и определяется эмпирический диапазон его применимости, что позволяет затем делать некоторые предположения о точности вычисления произвольного молекулярного свойства. Искусство выбора базисного набора основано на предшествующем опыте исследования родственных систем с использованием базисных наборов аналогичного качества. Задача выбора базисного набора для конкретного применения подробно обсуждается в разд. [26]
Следует отметить, что вычисляемый вклад в энергию, обусловленный переносом заряда, сильно зависит от качества исходной волновой функции и заметно снижается по мере ее уточнения, например по мере расширения базисного набора. В табл. 3 приведены компоненты AZ. Первые две строки содержат результаты расчета по теории возмущений [9] и по методу МО ЛКАО ССП [10] с использованием минимального базисного набора. В двух последних строках приведены данные расчетов по теории возмущений [11] и по методу МО ЛКАО ССП [5] с привлечением сравнительно широкого базисного набора; результаты последнего находятся в хорошем согласии с экспериментом. [27]
В 1960 - е годы введение базисов гауссовых функций для молекулярных расчетов ( основанное на предложении С. Ф. Бойза, сделанном в 1950 г.) значительно снизило вычислительное время, необходимое для получения хороших результатов при хартри-фоковских расчетах молекул, что сделало реальными расчеты больших молекул. Развиты и продолжают развиваться различные методы хотя бы частичной компенсации корреляционной ошибки. Полный расчет по методу конфигурационного взаимодействия с применением функций, определяемых выбранным базисным набором, в принципе должен исключить всю корреляционную ошибку, которую можно учесть при использовании данного базисного набора; однако проблема быстро становится практически неразрешимой при возрастании размеров системы. По этой причине расчеты по методу конфигурационного взаимодействия ( KB) проводятся лишь с учетом ограниченного числа конфигураций. В последнее время разработаны многоконфигурационные методы ССП, в которых волновые функции возбужденных конфигураций оптимизируются одновременно с оптимизацией функции основного состояния. Эти и многие другие усовершенствования призваны постоянно повышать точность молекулярных расчетов. Тем временем удается непрерывно получать полезные результаты с использованием уже отработанных методов. [28]
Если бы в каждом случае удалось разрешить свою секулярную проблему, то пришли бы в результате в точности к тем же приближенным энергиям и волновым функциям, хотя разложения в обоих случаях по внешнему виду совершенно различные. Причина такого совпадения просто в том, что каждый МО-детерминант может быть разложен ( при использовании МО в приближении ЛКАО) по соответствующим АО-детерминантам. Вообще говоря, функция-произведение молекулярных орбиталей представляется в виде линейной комбинации всех функций-произведений атомных орбиталей с соответствующими числовыми коэффициентами, и, следовательно ( учитывая спиновые множители и проводя антисимметризацию), разложенные МО-детерминанты, даже для одной МО-конфигурации, могут содержать все возможные АО-детерминанты. Поэтому произвольная МО-функция может быть разложена по полному набору всех АО-детерминантов, которые можно построить с использованием первоначального ограниченного базисного набора атомных орбиталей. Таким образом, данная многодетерминантная функция может быть представлена в двух эквивалентных формах по методу ВС и методу МО. Базисные детерминанты, используемые в методе МО и методе ВС, фактически связаны между собой соотношением ФФТ [ ср. [29]
Комбинация выбранных атомиых орбита-лей называется базисным набором. Включение дополнительных орбиталей в базисный набор приводит к расширенному базисному набору. Соображения экономии диктуют использование минимального базисного набора везде, где это возможно. [30]
Страницы: 1 2 3