Деструкция - боковая цепь
Cтраница 1
Деструкция сахароподобной боковой цепи до метильной группы, без сомнения, включает переносы гидрид-иона, в результате которых происходит восстановительное отщепление фенольной гидроксильной группы и превращение производного антрахинона в производное антрона. [1]
Кинетика деструкции боковых цепей, в том числе содержащих серу, и повторного их взаимодействия с углеродом или металло-органическими примесями с образованием новых, более стойких промежуточных соединений, имеет решающее значение при выборе оптимальных режимов одного из важнейших этапов процесса облагораживания нефтяного углерода - обессеривания, а также для изучения процесса графитации. [2]
В результате деструкции боковых цепей происходит двумерная укладка слоев, обусловливающая дальнейшие изменения физико-химических свойств углеродистого материала. Внутри гексагональных сеток кристаллитов кокса связи весьма прочны; при отсутствии химически активных реагентов они могут быть разрушены лишь с помощью высоких температур. [3]
Учет кинетики деструкции боковых цепей, в том числе содержащих серу, и повторного их взаимодействия с углеродом кокса или металлоорганическими примесями с образованием новых, более стойких промежуточных соединений, имеет решающее значение при выборе оптимальных режимов одного из важнейших этапов процесса деструктивных превращений нефтяного кокса - обессе-ривания, а также для изучения процесса графитации. [4]
Корреляция между интенсивностью процессов деструкции боковых цепей и образования: поперечных связей при облучении полиакрилатов наблюдается не всегда. Необходимо одновременно изучить оба процесса. Необходимо также исследование вопроса о влиянии подвижности сегментов макромолекулы на способность полиакрилатов и полиметакрилатов к образованию поперечных связей под действием ионизирующего излучения. Для полибутилакрилатов, различающихся строением бутиль-ного радикала, разность между комнатной температурой и температурами стеклования TKOWS - Гст возрастает в ряду трет О.т. гэр мзон-бу - тил. По эффективности процесса образования поперечных связей при комнатной температуре эти полимеры располагаются в обратном порядке. [5]
Алкилфенолы в условиях гетерогенного гидрогенизационного катализа вступают в реакции деалкилирования, деструкции боковой цепи, диспропорционирования, изомеризации, а также, в отличие от превращений в присутствии кислотных катализаторов, в реакции восстановления и гидрирования. Другими словами, эти катализаторы являются по отношению к фенолам электрофильными агентами. Следовательно, следует ожидать, что в случае гетерогенного гидрогенизационного катализа сохранятся высказанные выше основные положения, характеризующие влияние электронного строения на реакционную способность алкилфенолов в реакциях электро-фильного замещения. [6]
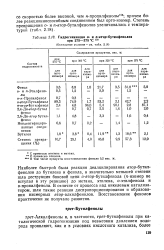 |
Гидрогенизация о - и п-вгор-бутилфенолов при 275 - 375 С 15в. [7] |
Наиболее быстрой была реакция деалкилирования втор-бутил-фенолов до бутилена и фенола, в значительно меньшей степени шла деструкция боковой цепи о-втор-бутилфенола ( n - изомер не вступал в эту реакцию) до метана, этилена, о-этилфенола и о-пропилфенола. [8]
Если мы предположим, что деструкция происходит преимущественно путем расщепления эфирных групп и со скоростью, сравнимой со скоростью деструкции боковых цепей в полиметил-метакрилате, то можно ожидать, что Ed составляет примерно 60 эв. Если предположить, что сшивание происходит таким же способом, как в полиакрилатах, то Ес можно оценить примерно в 90 эв. [9]
При увеличении размеров алкильных групп стерический фактор может возобладать над электронным, что видно, например, из сопоставления молекулярных диаграмм и экспериментальных данных в случае гидрогенизации трег-бутилфенолов, где не наблюдается деструкция боковой цепи. [10]
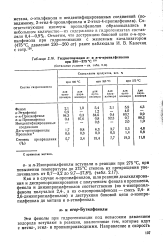 |
Гидрогенизация о - и п-м-пропилфенолов при 300 - 375 С 158. [11] |
Как и в случае н-пропилфенолов, шли реакции деалкилирова-ния и диспропорционирования с получением фенола и пропилена, фенола и диизопропилфенолов соответственно ( из п-изопропил-фенола получался 2 4 -, а из о-изопропилфенола - смесь 2 4 - и 2 6-диизопропилфенолов) и деструкция боковой цепи о-изопропилфенола до метана и о-этилфенола. [12]
Этилфенол не деалкилируется в принятых условиях, что объясняется, по-видимому, как и в реакциях диспропорционирования, его малой общей реакционной способностью, определяемой, как отмечено выше, трудностью образования я-комплекса. В реакции деалкилирования ( образование фенола и этана) и деструкции боковой цепи ( образование крезола и метана) о-этилфенол обладает большей активностью, чем пара-изомер, причем разрыв по а ( 3-связи боковой цепочки более предпочтителен. Эти данные находятся в соответствии со значениями электронной плотности на соответствующей связи, уменьшение которой благоприятствует рассматриваемым реакциям. [13]
При дегидрировании парафиновых углеводородов ZnCrO4 высокоселективен, так как, обладая способностью дегидрировать углеводороды, совсем не способен их циклизо-вать, чем заметно отличается от других дегидрирующих катализаторов. При температурах несколько ниже 600 С происходит гладкое дегидрирование этилбензола в стирол. При 600 С наблюдается уже деструкция боковой цепи с образованием бензола и толуола; при дальнейшем повышении температуры деструкция задевает и бензольное кольцо, причем при температуре около 700 С этот процесс достигает значительной глубины. Применение в качестве носителей ZnO или Сг2О3 не повышает дегидрирующих свойств хромата цинка, способствуя, наоборот, некоторому увеличению крекинга боковой цепи. [14]
В этих условиях главным направлением в превращении алкилфенолов является диспропорционирование и деалкилирование ( для первичных и вторичных алкилфенолов шла также деструкция боковой цепи), причем с увеличением длины и степени разветвленности алкильной группы повышается реакционная способность соединений и значение реакции деалки-лирования в общем превращении. [15]
Страницы: 1 2