Химически чистая азотная кислота
Cтраница 3
 |
Нормы качества дистиллированной воды ( или конденсата пара. [31] |
Анализ производится следующим образом. В коническую колбу емкостью 100 мл берут 25 мл раствора 1: 9 аккумуляторной кислоты или 1: 4 раствора электролита, добавляют 1 мл раствора химически чистой азотной кислоты удельного веса 1 15 или же 5 - 6 кристаллов химически чистого персульфата аммония и нагревают до кипения. После охлаждения до комнатной температуры к раствору прибавляют 1 мл 10 % - ного раствора K4Fe ( CN) 8 и взбалтывают. Если при этом получится зеленоватое или очень слабое голубоватое окрашивание раствора, то кислота и электролит годны. [32]
Остальные соли растворяют таким же методом в водном растворе цианида калня и вводят в ванну. Применяемый в качестве соли нитрат серебра растворяют в дистиллированной воде в темном помещении Отдельно готовят раствор цианида калия и приливают его при перемешивании к раствору нитрата серебра Образующийся белый осадок растворяется в избытке цианида калия При отсутствии солей серебра можно приготовить электролит анодным растворением серебра в водном растворе цианида катая или, используя нитрат серебра, полученный растворением металлического серебра в концентрированной химически чистой азотной кислоте. [33]
В соответствии с ГОСТ 4658 - 49 для суммарного количественного определения примесей в ртути берут 100 г ртути марки Р1 или 50 г ртути марки Р2, помещают в небольшую колбу или реторту и отгоняют на умеренном пламени горелки, просасывая воздух и собирая отгон в приемник. Отгонку ведут до тех пор, пока в дистилляторе останется не более 1 - 2 г ртути. Остаток растворяют в небольшом количестве химически чистой азотной кислоты ( плотность 1 4 г / см3), раствор сливают во взвешенный фарфоровый тигель, емкостью 15 мл, выпаривают, сушат и при 500 С осторожно прокаливают до постоянной массы. По разности масс пустого тигля и тигля с остатком находят массу нелетучего остатка, по которой судят о чистоте ртути. [34]
В соответствии с ГОСТ 4658 - 49 для суммарного количественного определения примесей в ртути берут 100 г ртути марки Р1 или 50 г ртути марки Р2, помещают в небольшую колбу или реторту и отгоняют на умеренном пламени горелки, просасывая воздух и собирая отгон в приемник. Отгонку ведут до тех пор, пока в дистилляторе останется не более 1 - 2 г ртути. Остаток растворяют в небольшом количестве химически чистой азотной кислоты ( плотность 1 4г / сж3), раствор сливают во взвешенный фарфоровый тигель, емкостью 15 мл, выпаривают, сушат и при 500 С осторожно прокаливают до постоянной массы. По разности масс пустого тигля и тигля с остатком находят массу нелетучего остатка, по которой судят о чистоте ртути. [35]
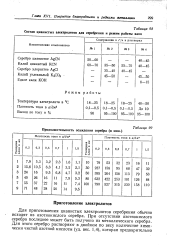 |
Состав цианистых электролитов для серебрения и режим работы ванн. [36] |
Для приготовления цианистых электролитов серебрения обычно исходят из азотнокислого серебра. При отсутствии азотнокислого серебра последнее может быть получено из металлического серебра. Для этого серебро растворяют в двойном по весу количестве химически чистой азотной кислоты ( уд. [37]
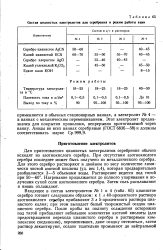 |
Состав цианистых электролитов для серебрения и режим работы ванн. [38] |
Для приготовления цианистых электролитов серебрения обычно исходят из азотнокислого серебра. При отсутствии азотнокислого серебра последнее может быть получено из металлического серебра. Для этого серебро растворяют в двойном по весу количестве химически чистой азотной кислоты ( уд. Нагревание продолжается до полного упаривания и получения сухой соли азотнокислого серебра. Затем соль расплавляют и вновь охлаждают. [39]
По Hefi y содержание золы определяют, осторожно сжигая 5 г нитроклетчатки с небольшим количеством чистого парафина, содержание золы в котором должно быть предварительно определено таким же способом; сжигание производят в платиновой чашке или в платиновом тигле или же в фарфоровом тигле. Золу смачивают раствором углекислого аммония и несколько раз нагревают при 200 до постоянного веса. Можно также 5 г нитроклетчатки смочить в платиновой чашке 25 мл химически чистой азотной кислоты ( плотн. По В е г Гю нитроклетчатку обливают в фарфоровом тигле сернистым аммонием, не содержащим золы, выпаривают досуха на водяной бане и прокаливают. Хорошая нитроклетчатка, изготовленная из хлопка, содержит обычно менее 0 5 / 0 золы. [40]
В коническую колбу отмеривают пипеткой 25 мл титрованного раствора AgNO3, подкисленного химически чистой азотной кислотой, прибавляют 2 - 3 мл раствора железо-аммонийных квасцов ( индикатор), подставляют колбу под бюретку, подложив белый лист бумаги, и титруют роданидом аммония до красноватого окрашивания. Раствор NH4CNS приливают из бюретки медленно, по каплям, все время сильно взбалтывая жидкость в колбе. При этих условиях образующееся роданистое серебро будет свертываться и быстро оседать на дно колбы, поэтому жидкость над осадком будет более прозрачной и в ней затем легче будет наблюдать появление красного окрашивания. [41]
На технических весах отвешивают 8 г чистого сухого NH4CNS ( KCNS - около 10 г) и растворяют в 1 л воды. Для установки нормальности роданида аммония можно воспользоваться титрованным раствором AgNOs - Поступают так. В коническую колбу отмеривают пипеткой 25 мл титрованного раствора AgNO3, подкисленного химически чистой азотной кислотой, прибавляют 2 - 3 мл раствора железо-аммиачных квасцов ( индикатор), подставляют колбу под бюретку, подложив лист белой бумаги, и титруют раствором роданида аммония до красноватого окрашивания. Раствор NH4CNS приливают из бюретки медленно, по каплям, все время сильно взбалтывая жидкость в колбе. Образующийся роданид серебра свертывается и быстро оседает на дно колбы; поэтому жидкость над осадком будет более прозрачной и в ней легче наблюдать появление красного окрашивания. Титрование считают оконченным, когда от одной капли появляется красноватое окрашивание раствора, не исчезающее при сильном взбалтывании. [42]
 |
Стандартная серия растворов для приготовления перлов. [43] |
Вследствие высокой чувствительности метода требуется особая тщательность при выполнении анализа. Все применяемые реактивы и посуда должны быть проверены в фильтрованном ультрафиолетовом свете на отсутствие флуоресценции. Воздушная среда лаборатории, где производят анализ, и рабочее место не должны быть загрязнены соединениями урана. Платиновые петли тщательно очищают от соединений урана. Видимые остатки плавня удаляют механически, затем платиновую петлю накаливают докрасна в окислительном пламени горелки и погружают на 2 - 3 сек в концентрированную химически чистую азотную кислоту и снова прокаливают. Таким образом петлю обрабатывают несколько раз и проверяют на отсутствие флуоресценции. Фильтры должны быть проверены на отсутствие флуоресценции и гасителей флуоресценции. Для этого их обрабатывают в аналогичных условиях, как и пробу. [44]
Учитывая, что флуоресцентный метод анализа обладает высокой чувствительностью, необходимо соблюдать особую тщательность и предосторожность при выполнении анализа. Все применяемые реактивы и посуда должны быть проверены в фильтрованном ультрафиолетовом свете на отсутствие флуоресценции. Воздушная среда лаборатории не должна быть загрязнена соединениями урана. На рабочем столе, где производят анализ проб и готовят стандартные перлы, нельзя одновременно проводить другой работы с соединениями урана. Необходимо следить за тем, чтобы платиновая петля не была загрязнена соединениями урана. Видимые остатки плава фторида натрия удаляют механически. Для полной очистки петли от металла ее накаливают докрасна в окислительном пламени горелки и погружают на 2 - 3 сек в концентрированную химически чистую азотную кислоту и снова прокаливают. Петлю обрабатывают таким образом несколько раз и проверяют на отсутствие флуоресценции. Фильтрующие материалы должны быть проверены на отсутствие флуоресценции, для чего их обрабатывают в аналогичных условиях, как и анализируемую пробу; они должны быть также проверены на отсутствие гасителей флуоресценции. Для этой цели определенное количество стандартного раствора наносят на фильтры и обрабатывают как и пробу. Интенсивность свечения полученного при этом перла сравнивают со стандартным перлом, содержащим такую же концентрацию урана. [45]
Страницы: 1 2 3