Неводный компонент
Cтраница 2
Исходя из представлений об одновременном наличии сольватирующего-и дезассоциирующего типа взаимодействий ( в указанном выше расширенном смысле) между неводным компонентом бинарного растворителя и химически более близким к нему, чем к воде, дополнительным компонентом, повышение растворимости соли в водном этаноле при добавке бензол а можно объяснить и не допуская, что сольватация спирта бензолом является единственным результатом введения дополнительного компонента в систему. Для этого достаточно предположить, что при данных условиях она является лишь преобладающим процессом. [16]
Учитывая, что для гидрофобных эффектов характерна сильная температурная зависимость и что механизм растворения аргона связывается с образованием квазиклат-ратных структур вокруг молекул неводного компонента или ее гидрофобных радикалов [56, 59, 62], можно заключить, что это же явление имеет место и вблизи СН3 - группы метанола. [17]
При испарении воды из раствора по мере повышения его концентрации изменяется удельный расход теплоты в связи с изменением характера взаимодействия между водой и неводными компонентами. [18]
В данной главе последовательно рассматриваются парциальные молярные объемы компонентов в жидкой воде, в жидкой неводной фазе, в кристаллогидратной фазе, в газовой смеси неводных компонентов и водяного пара. [19]
Полученные результаты не укладываются в прежнее объяснение влияния дополнительного компонента на растворимость веществ в бинарных растворителях, которым учитывалось только взаимодействие сольватационно-го типа между дополнительным компонентом и неводным компонентом бинарного растворителя. Наоборот, предложенная нами схема, учитывающая взаимное наложение взаимодействий двух типов, которые влияют противоположным образом на растворимость, удовлетворительно объясняет как положительный, так и отрицательный знак эффекта дополнительного компонента и случаи его инверсии. [20]
Эти неводные компоненты ( метан, азот и диоксид углерода) иногда встречаются в смесях в достаточно больших и сопоставимых концентрациях, поэтому ни один из этих компонентов нельзя рассматривать как основной неводный компонент. [21]
Здесь Т и Г - температуры ( в К) испарения растворителя из раствора и чистого растворителя при давлении Р; Р и Р - давление ( в Па) пара над чистым растворителем при температуре Т и давление, при котором ведется испарение растворителя из раствора; хп и х - концентрация неводного компонента в паровой фазе и в растворе, мол. [22]
С увеличением концентрации органического компонента электролит сильнее всаливается в фазу ионита. Линейное возрастание концентрации неводного компонента вызывает экспоненциальное возрастание коэффициента распределения, в то время как в элюентной хроматографии увеличение концентрации элюента уменьшает величину коэффициента распределения. [23]
При адсорбции на металле анионы раствора могут конкурировать с непредельными соединениями. Показано, что с ростом концентрации неводного компонента в кислых водно-этиленгликолевых растворах ингибирующее действие пропаргилового спирта увеличивается. Это говорит об участии в адсорбционно-хи-мических превращениях ацетиленовых ингибиторов не только собственно поверхности металла, но и адсорбированных на ней молекул воды или анионов гидроксида, что подтверждается усилением ингибирующего действия пропаргилового спирта, гексинола и других ацетиленовых соединений с концевой тройной связью при добавлении в кислые растворы галогенид-ионов. [24]
Составы природных систем, содержащих воду, обычно подчиняются определенным закономерностям. Часто встречаются системы, в которых один неводный компонент является основным, а множество других компонентов присутствуют в виде малых примесей. В качестве таких основных компонентов обычно выступают метан, азот, диоксид углерода. Многокомпонентные системы такого типа по своему фазовому поведению мало отличаются от соответствующих двойных систем с водой. [25]
Для этого случая в области не очень высоких температур содержание неводного компонента в воде мало. [26]
Для выяснения степени влияния гидратной оболочки на величину сорбции, как нами уже указывалось, были проведены опыты в спиртоводной и диоксано-водной средах. Как видно из полученных данных ( см. рис. 1), добавление неводного компонента к раствору изменяет сольват-ную оболочку; так, при 25 величина десорбции кальция в водной среде в 2 раза больше, чем в диоксановодной и намного больше, чем в спиртоводной, и с повышением температуры процесс десолыватации идет значительно слабее, чем дегидратации. Последнее подтверждается постоянством величины изменения свободной энергии при обмене в смешанных растворителях. F для обмена эспатита кальция на литий в спиртоводной среде в интервале температур 25 - 50 равно 2679 кал, а для этого же случая обмена в диоксановодной среде - AF2690 кал. Из данных обмена эспатита кальция на ионы калия и лития ( табл. 1) видно, что до 50 заметных изменений в дегидратации лития не наблюдается, так как десорбция кальция калием составляет 103 мг-экв на 100 г сорбента, а литием в этих условиях - всего 63 мг-экв. Так, при 95 десорбция кальция литием достигает уже 120 мг-экв, а калием - только 115 мг-экв на 100 г сорбента. [27]
Причины изменения устойчивости жидкофазных систем необходимо анализировать с учетом термодинамических свойств всех участников равновесия. В работе [51], в частности, приведены доказательства недостаточности оценки лишь концентраций воды и неводного компонента как реактантов. В случае реакции комплексообразования добавление к воде этанола приводит к росту отрицательных значений А5 реакции. Существенную роль в этом играет процесс сольватации лиганда. Большее влияние изменения состава жидкой фазы на свободные энергии переноса иона № 2 по сравнению с комплексными ионами должно было бы приводить к снижению устойчивости комплексов при повышении концентрации спирта в воде. Однако наблюдаемый рост устойчивости обусловлен уменьшением AG сольватации аммиака. Повышение устойчивости комплексов на практике усложняет процесс удаления тяжелого металла из жидкофазной системы. [28]
При исследовании тройных водных систем даже и при частичной кристаллизации пробы применялся иногда тип автоклава, изображенный на рис. 20, а. В этом случае отобранная проба извлекалась водой и в полученном растворе путем анализа устанавливалось отношение количеств неводных компонентов, содержание же воды в отобранных пробах рассчитывалось по соотношению количеств загруженных в автоклав воды и того неводного компонента ( соли или едкой щелочи), который, как должно быть заведомо известно, в твердую фазу не переходит ни в виде индивидуальных кристаллов, ни в виде химического соединения или твердого раствора с другим компонентом. При этом делалась поправка на количество воды, перешедшее внутри автоклава в паровую фазу. [29]
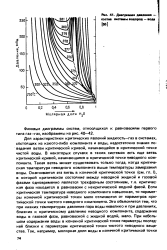 |
Диаграмма давления - состав системы водород - вода. [30] |
Страницы: 1 2 3 4