Описание - электронная структура
Cтраница 3
Однако не может быть передачи и сложения энергии у несуществующих материальных объектов. Канонические резонансные структуры являются мысленными, а не реальными образами, а сам резонанс - это только математический способ описания электронной структуры сопряженной молекулы с помощью подходящих валентных схем. Большинство резонансных структур, в том числе биполярные ионные структуры, не могут возникнуть даже в условиях оптического возбуждения, когда поглощается энергия в сотни кДж / моль. [31]
Заселение атомных орбиталеи электронами происходит согласно принципу заполнения и принципу Паули. Атомные орбитали используются не только для описания электронной структуры атомов, но и при исследованиях молекул: см. разд. Молекулярные орбитали и Линейные комбинации атомных орбиталеи. [32]
Теоретическое описание процессов хемосорбции опирается на многие разделы физики и химии. В образовании химической связи между адсорбированным атомом ( или молекулой) и поверхностью твердого тела принимают участие наиболее слабосвязанные электроны системы. Поэтому необходимой и важной частью теории должно быть описание электронной структуры атомов и молекул, находящихся близко к поверхности. [33]
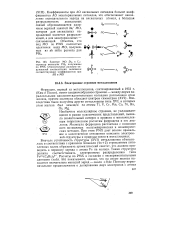 |
Занятые МО D3h и C4v структур молекулы PHs, полученные по РМХ. Относительные размеры окружностей соответствуют величинам соответствующих коэффициентов при АО в МО. [34] |
Необычное молекулярное строение, не укладывающееся в рамки классических представлений, вызвало значительный интерес и привело к многочисленным теоретическим расчетам ферроцена и его аналогов. Молекула ферроцена рассчитана с помощью всех возможных полуэмпирических и неэмпирических методов. При этом РМХ дает вполне правильное в качественном отношении описание электронной структуры и природы связи в металлоценах. Вначале устойчивость структуры ( XVI) металлоценов объяснялась в соответствии с правилом Хюккеля ( 4п 2) стремлением пяти-членных колец образовать ароматический секстет, что должно приводить к переносу заряда с атома Fe на кольца. [35]
В результате решения уравнений Хартри - Фока находят некоторую систему канонических орбиталей. Химические процессы мыслятся большей частью в терминах разрыва одних и формирования других химических связей. В связи с этим исходная информация о молекулярных орбиталях может быть преобразована в новую с тем расчетом, чтобы описание электронной структуры бьшо дано в терминах локализованных орбиталей. При этом для определенного класса молекулярных систем теоретически удается установить некоторые характеристики отдельной связи, такие, как дипольный момент, продольная и поперечная поляризуемости и др. В методе МО не вводят априорные понятия о кратности связей. Тем не менее после завершения решения уравнений Хартри - Фока могут быть найдены величины, которые коррелируют со сложившимися представлениями о кратности в рамках представлений о спин-валентности. [36]
Трудности, возникающие при построении теории электронной структуры кристаллов с ЛЦ, связаны с необычной природой самих объектов. С одной стороны, они представляют собой системы молекулярного типа, так как трансляционная симметрия, присущая идеальным кристаллам, при появлении дефекта нарушается: как и у молекул, у кристаллов с ЛЦ группа симметрии точечная. Поэтому оказываются неприменимыми традиционные методы, развитые в зонной теории твердых тел и с успехом применявшиеся уже с конца тридцатых годов для описания электронной структуры идеальных кристаллов. Для совершенного кристалла благодаря наличию трансляционной симметрии порядок вековых уравнений, решаемых при расчете электронных состояний ( по крайней мере для валентных зон), определяется числом атомов в примитивной ячейке. [37]
По существу дилемма локализация - делокализация возникла в период становления квантовой химии как самостоятельного раздела науки. Если метод ВС основан на концепции двухэлектронных локализованных связей, которая соответствует обширным экспериментальным данным структурной химии о локализованном характере обычных химических связей, то метод МО, представляющий собой распространение оболочечной модели атома на случай молекулы, приводит к описанию электронной структуры в терминах делокализованных одноэлектронных волновых функций. Впоследствии выявилась ограниченность обоих подходов; последующее их уточнение сближало получаемые результаты и их физическую интерпретацию. Действительно, учет резонансных и ионных структур вводит в метод ВС элементы делокализации, а учет конфигурационного взаимодействия ( KB) в методе МО приводит к более локализованным электронным структурам. [38]
Метод функционала плотности ( МФП) энергии ( термодинамического потенциала) является в настоящее время наиболее эффективным методом описания сильновзаимодействующих плотных систем. Последовательное применение МФП для описания свойств плотной электрон-ионной системы сочетает простоту квазиклассического метода с аккуратным учетом обменно-корреляционных эффектов и выделением связанных состояний. Однако здесь отсутствуют большие математические сложности, которые содержит последовательное использование метода Хартри-Фока. Не удивительно, что МФП и его модификации были с успехом использованы в различных областях физики для описания электронной структуры атомов и молекул, кластеров и иных малых частиц, поверхностных явлений, свойств твердых и жидких металлов, неидеальной плазмы и др. МФП хорошо приспособлен для исследования уравнений состояния плотных систем. [39]
Тот факт, что два противоположных представления могут быть использованы для развития расчетных методов, подчеркивает гибкость квантовой химии. Однако при описании строения молекул подобная двойственность неоднократно приводила к недоразумениям. Это зависит прежде всего от мнения исследователя и соображении практического удобства. Однако на основе этих подходов могут быть развиты два типа приближений, отличающихся методами расчета, но приводящих к единому результату - описанию полной электронной структуры. При этом начальные приближения могут быть даже эквивалентны математически, как в случае применения теории молекулярных орбиталей ( МО) для основных состояний с закрытой оболочкой, описываемых двумя отличающимися фазовым множителем слейтеровскими детерминантами, построенными из делокализованных или локализованных МО. [40]
В квантовой химии молекул разработаны достаточно простые полуэмпирические схемы расчета, основанные на приближении МО ЛКАО, их возможности для молекул хорошо изучены. Конечно, при распространении эти схем на кристалл специального рассмотрения требует вопрос о калибровке параметров, предложенной для молекул - в случае кристалла молекулярные параметры должны быть, вообще говоря, модифицированы. Поэтому при рассмотрении твердых тел предпочтительнее те расчетные схемы, в которых используются, по возможности, лишь атомные характеристики - потенциалы ионизации, волновые функции и др. Примером подобного рода расчетной схемы, полезной главным образом для ионных кристаллов, является метод Малликена-Рюденберга. Результаты ряда расчетов, обсуждавшихся в предыдущей главе, подтверждает это соображение. Специальную задачу представляет собой учет влияния поля кристалла на выделенную подсистему - дефект и его окружение. Применение молекулярных моделей в теории кристаллов с локальными центрами сопряжено с решением более общей задачи - описания электронной структуры твердых тел исходя из свойств образующих их атомов. Такая задача является основной в квантовой химии твердого тела. [41]
Интересно отметить, что большой разброс результатов, найденных в гл. Сложность состоит в том, что d - co - стояния заполнены и заполнены ( в том смысле, как это бывает ь ионных кристаллах) также 5 - и / - состояния соседних ионов галогена. При этом понижение энергии первых компенсируется повышением энергии вторых, и явления химического захвата в этом случае нет. Вместо этого наиболее важный вклад возникает из-за деформации ионов ( см. гл. При таком рассмотрении полной энергии первый важный шаг состоит в выборе тех атомных орбиталей, которые необходимы для описания электронной структуры, и в выяснении того, какие из этих орбиталей заполнены, Только после этого можно воспользоваться теорией возмущений и найти соответствующее изменение полной энергии. [42]
Страницы: 1 2 3