Остаток - гистидин
Cтраница 1
Остаток гистидина в миоглобине и гемоглобине несколько смещен относительно правильного шестого координационного положения иона железа. В некоторых аномальных гемоглоби-нах человека этот остаток оказывается замещенным. В НЬМ, где он замещен тирозином, железо окисляется до ферри-иона и способность железа служить переносчиком кислорода утрачивается. Замещение гистидина аргинином не приводит к утрате способности переносить кислород. Считая, что остаток тирозина может образовывать комплекс с железом, объясните большее сродство фенольных групп к Fe3, чем у гистидина и аргинина. [1]
В активном центре РНК-азы находится два остатка гистидина 12 и 119, осуществляющие катализ по типу общего кислотного катализа. [2]
Предложен метол тверлофаэяого синтеза пептида, в котором остаток гистидина связан со смолой таким образом, что полипептнлная цепь может наращиваться и но амино -, н по карбоксильной группам гистидина, прячем ни одна нз этих групп со смолой непосредственно не связана. Последовательность реакций изображена ниже; Приведите уравнения, показывающие, какие реакции происходят на каждой стадии. [3]
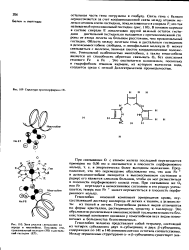 |
Структура протопорфирина IX.| Зона участка связывания кл лоро да в миоглоби не. Пока за ны гем, проксимальный гистидин ( F8 и днстальный гистидин ( Е7. [4] |
В гемовом кармане в составе спирали Е локализован другой важный остаток гистидина днстальный гистидин; он находится с противоположной сто роны от атома железа на большем расстоянии, чем проксимальный гистидин. Область между железом гема н днстальным гистидином в дезоксимиоглобине свободна, н лилофильная молекула О может связываться с железом, занимая шестое координационное положение. Это оказывается возможным, поскольку в гидрофобном гемовом кармане, из которого вытеснена вода, создается среда с низкой Диэлектрической проницаемостью. [5]
Донором протонов, вероятно, является имидазольное кольцо остатка гистидина, связывающее ионы металлов. [6]
По имеющимся данным, в состав активного центра аспартатаминотрансферазы входит остаток гистидина. [7]
Естественно, что ни в существенно кислой среде, когда оба остатка гистидина протонированы, ни в существенно щелочной среде, когда оба остатка лишены протонов, каталитическое превращение осуществляться не будет. Процесс возможен лишь в некоторой промежуточной области, в которой содержание монопротонированной формы достаточно велико. [8]
На основании этих результатов Брюс [118] предположил, что имидазольная группа остатка гистидина может входить в состав каталитического центра ферментов, ответственных за специфический гидролиз тноэфиров. [9]
Более того, как показало исследование кинетики, в действии протеаз принимает участие остаток гистидина. Поскольку в молекуле трипсина гистидин не примыкает непосредственно к активному серину, приходится предположить, что пептидная цепь фермента свернута таким образом, что остаток гистидина уже вторично оказывается расположенным рядом с активным серином. [10]
Изучение влияния рН на скорость катализируемых химотрипсином реакций свидетельствует о необходимости для катализа непротонированного остатка гистидина или его кинетического эквивалента. [11]
Гидролиз специфических сложноэфирных субстратов, катализируемый а-химотрипсином, протекает при участии имидазоль-ной группы остатка гистидина активного центра фермента, выступающей в качестве общекислотно-общеосновного катализатора. [12]
Катализ сериновыми протеазами включает стадию, катализируемую общим основанием, роль которого выполняет имидазолильная группировка остатка гистидина, поэтому соединение 10.14 можно рассматривать как модель ацилфермента. [13]
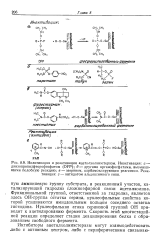
Функциональной группой, ответственной за гидролиз, является здесь ОН-группа остатка серина, нуклеофильные свойства которой усиливаются имидазольным кольцом соседнего остатка гистидина. Нуклеофильная атака сериновой группой ОН приводит к ацетилированию фермента. Скорость этой многостадийной реакции определяет стадия деацилирования белка с образованием свободного фермента. [15]
Страницы: 1 2 3 4