Перемещение - заместитель
Cтраница 2
Показано, что увеличение расстояния между хинуклидиновым ядром и дифенилкарбинольным остатком за счет введения между ними метиленовых групп, а также уменьшение этого расстояния путем перемещения заместителя в положение 2 хинуклидинового ядра ( XXVIII), частичное дегидрирование хинуклидинового фрагмента - в положениях 2, 3, а также переход от третичных спиртов ко вторичным ( XXVIII, R H) значительно снижают антигистаминную активность. Неактивны вещества, в которых хинуклидино-вое ядро замещено другими гетероциклическими остатками, значительно убывает антиаллергическая активность при R1 и К2 - алкил независимо от длины алкильных цепей. Интересно, что переход от ( хинуклидил - З) дигексилкарби-нола ( XXVII, К1 Н2СбН1з) к его циклогексильному аналогу повышает антиаллергическое действие вещества. Введение в арильные остатки R1 и R2 мета - и пара-заместителей понижает, ортозаместителей усиливает активность. [16]
Было обращено внимание на то, что в этих системах возможны два различных способа циклизации: при образовании новой связи между концевыми ( в сопряженной системе) атомами С у каждого из этих атомов происходит такое перемещение заместителей, что один из них оказывается над плоскостью цикла, другой - под плоскостью. Другими словами, при таком перемещении группы СН3 в 1 4-диметил-бутадиене либо окажутся одновременно над плоскостью цикла, либо одна - над плоскостью цикла, другая - под ней. [17]
Так, например, исходя из этой формулы, нельзя понять образования орсина ( 325) из неароматической части молекулы гризеофульвина, если одновременно не принять, что в процессе его сплавления со щелочью имеет место перемещение заместителей. Последнее же, однако, до сих пор доказано не было. Поэтому вместо формулы ( 326) несколько позднее1400 была предложена другая структурная формула гризеофульвина ( 321), которая кажется более вероятной, хотя и она должна рассматриваться как не вполне обоснованная. [18]
В результате отщепления X со своими связующими электронами на о-атоме углерода образуется секстет электронов. Это вызывает перемещение заместителя от р - к а-углеродному атому; заместитель также переходит со своими связующими электронами. [19]
Как было указано Гаттерманом [4], алкилбензолы более легко вступают в реакцию синтеза альдегидов по Гаттерманну-Коху, чем сам бензол, причем вводится только одна альдегидная группа в пар-положение но отношению к присутствующей алкильной группе. Если аса-положение занято, то может происходить перемещение алкилыюго заместителя, находящегося в гааоа-положении. Хардинг и Коен [19] сообщили об образовании 2 5-диметилбензальдегида из л-ксилола. [20]
Целесообразность ознакомления с данными по межмолекулярному перераспределению алкильных групп в рамках данной книги обусловлена тем обстоятельством, что этот процесс имеет много общего с изомер ными превращениями алкилароматических соединений. Кроме того, знание основных закономерностей реакций, связанных с межмолекулярным перемещением заместителей, облегчает нахождение таких условий изомеризации, в которых побочное образование продуктов диспропорционирования невелико. [21]
Нафтеновые углеводороды в условиях каталитического крекинга расщепляются с разрывом кольца и образованием изопарафинов. Второй важной реакцией нафтенов является специфичная для каталитических процессов изомеризация структуры кольца ( обычно шестичленного в пятичленное) и перемещение заместителей. [22]
Использование вместо соляной кислоты безводной р-нафталинсуль-фокислоты позволило осуществить изомеризацию алкил - ( 1-хлорнаф-тил - 8) - сульфонов при значительно более низкой температуре. Причину ускорения процесса изомеризации при переходе от концентрированной соляной кислоты к безводной р-наф-талинсульфокислоте легко понять, если учесть, что перемещение заместителя в ароматическом соединении является следствием атаки протоном атома углерода, связанного с этим заместителем. В водном растворе кислоты условия для такой атаки малоблагоприятны, так как ароматическому соединению приходится конкурировать за обладание протоном с более основными молекулами воды. Количественную характеристику влияния кислотности среды на скорость реакций, катализируемых донорами протонов, дает функция кислотности Гаммета ( Я0), отражающая склонность среды к передаче протона нейтральному основанию. [23]
Соли цинка, кадмия и ртути широко используются для ускорения ионных процессов. Активность хлористого цинка в реакциях изомеризации парафинов или олефинов в изосоеди-нения [343-346], перемещения кратных связей в олефинах [356, 357] и перемещения алкиль-ных заместителей в ароматических соединениях [347, 348] в общем ниже, чем активность хлористого алюминия. [24]
Скорость полимеризации дивинила, его производных и некоторых непредельных олефиновых углеводородов и их производных зависит от характера, числа и положения заместителей в цепи. Для дивинила и его гомологов эти вопросы были выяснены С. В. Лебедевым [40], который пришел к следующим выводам: 1) при перемещении заместителей от крайних атомов сопряженной системы к средним скорость полимеризации в рядах изомеров возрастает, при обратном перемещении-убывает; 2) циклизация цепи, имеющей сопряженную систему двойных связей, повышает скорость полимеризации; 3) увеличение в гомологическом ряду массы заместителей у средних атомов сопряженной системы повышает скорость полимеризации, если нагревание вести при соответствующих температурах. Эти выводы, проверенные рядом других исследователей, оказались правильными. [25]
В разделе I Алканы ( парафиновые углеводороды) изомеры расположены в порядке уменьшения числа углеродных атомов в наиболее длинной углеродной цепи. Изомеры с равным числом углеродных атомов в наиболее длинной углеродной цепи размещены по мере увеличения числа замещающих алкилов; в случае одинакового числа заместителей изомеры расположены в порядке перемещения заместителей слева направо и усложнения их строения. [26]
В биокаталитических реакциях участвуют только 4 - и 5-оксиметильные группы; первая превращается в альдегидную группу пиридоксаля или в ами-нометильную группу пиридоксамина, а вторая этерифицируется фосфорной кислотой в пиридоксаль - 5а - фосфат или пиридоксамин - 5а - фосфат. Замещение гидроксильной группы в положении 5а водородом [ 11 J или аминогруппой, метилирование фенольного гидроксила [2, 200], образование N-метил-производного [ 111, замещение гидроксила в положении 4а водородом ( 4-де-зоксипиридоксин) [200] или одновременное замещение гидроксила в положениях 4а и 5а ( 4 5-быс-дезоксипиридокснн, CLVI) [200], перенесение гидроксильной группы из положения 3 в положение 6 ( при наличии в положении 5 аминометильной группы) [2 13] устраняют полностью или почти полностью витаминную активность, характерную для пиридоксина. Перемещение заместителей между различными положениями молекулы витаминов группы В6 лишает эти соединения физиологических свойств. Изопиридок-син ( CLVII), изопиридоксаль ( CLVIII) [214] и изопиридоксамин ( CLIX) не активны для высших организмов. [27]
Разумеется, такой способ определения дает правильные результаты лишь при условии, что превращения происходят без перемещения замещающих групп в бензольном ядре. При громадном большинстве реакций замещающие группы, действительно, сохраняют свое положение. Перемещение заместителей в ядре происходит лишь в редких случаях, которые в настоящее время достаточно хорошо изучены. Так, известно, что при сплавлении сульфокислот фенолов и галоидсульфокислот со щелочами при высокой температуре окси-группы из орто - и пара-положений могут перемещаться в мета-положение ( стр. [28]
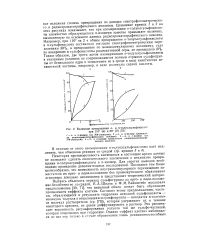 |
Взаимные превращения о - и п-толуолсульфокислот при 110 ( а и 80 ( б -. [29] |
Некоторая противоречивость имеющихся в настоящее время данных не позволяет сделать окончательного заключения о механизме превращения о-толуолсульфокислоты в - изомер. Для строгих выводов необходимо проведение дополнительных исследований. Последнее тем более целесообразно, что возможность внутримолекулярного перемещения заместителя из орто - в пара-положение без промежуточного образования ж-изомера довольно неожиданна и представляет теоретический интерес. [30]
Страницы: 1 2 3