Атом-атомный потенциал
Cтраница 2
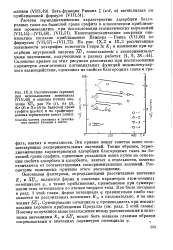
ДС / i, являются форма и значения параметров атом-атомных потенциалов ф, а также приближения, применяемые при суммировании этих потенциалов по атомам твердого тела. Как было показано в разд. KI, а следовательно, и на значениях Ai / i - Суммирование потенциала ф производилось при использовании хорошего приближения Крауэлла ( см. разд. [17]
Представляется перспективным, на наш взгляд, использовать метод атом-атомных потенциалов при прогнозировании катализаторов в такой бурно развивающейся области катализа, как катализ на цеолитах. Действительно, с помощью этого метода, в принципе, можно рассчитывать конформации органических молекул в микропорах цеолитов, диффузионные барьеры, оценивать свойства цеолитов как молекулярных сит. [18]
В работе [93] было предложено проводить подгонку параметров в атом-атомном потенциале типа Клементи ( ехр - 6 - 1) в два этапа. Вначале подгонка проводится по значениям энергии взаимодействия, вычисленным в первом и втором порядках теории возмущений. [19]
Выбор правильной симметрии кристалла часто оказывается за пределами точности метода атом-атомных потенциалов. Так, моноклинная модификация бензола, согласно атом-атомной схеме, получается при всех давлениях более устойчивой, чем ромбическая, в то время как в эксперименте моноклинная фаза наблюдается лишь при давлениях свыше 12 кбар. [20]
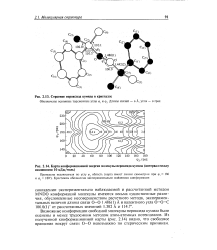 |
Строение пероксида кумила в кристалле.| Карта конформационной энергии молекулы пероксида кумила ( интервал между изолиниями 10 кДж / моль. [21] |
Возможные конформации свободной молекулы пероксида кумила были оценены и менее трудоемким методом атом-атомных потенциалов. Из полученной конформационной карты ( рис. 2.14) видно, что свободное вращение вокруг связи О-О невозможно по стерическим причинам. [22]
Если энергетические поверхности в случае Д рассчитывались в упомянутых выше работах методом атом-атомных потенциалов, то в ситуации X интересные результаты были получены квантово-химическим расчетом. [23]
В работе258 выполнено численное моделирование кристалла Сбо методом молекулярной динамики с использованием эмпирических атом-атомных потенциалов Леннард-Джонса с параметрами для графита. [24]
В последние годы для описания взаимодействия между двух-и многоатомными молекулами широко пользуются атом-атомными потенциалами. Взаимодействия между валентно не связанными атомами описывают сферически симметричными потенциалами ( обычно это потенциалы Леннард-Джонса или ехр - 6), и потенциальную энергию системы представляют как сумму таких потенциалов. [25]
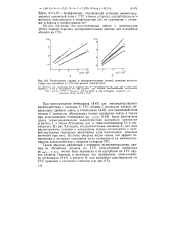 |
Рассчитанные ( линии и экспериментальные ( точки значения констант Генри для адсорбции на ГТС при разных температурах. [26] |
Таким образом, найденный с помощью экспериментальных данных по абсорбции этилена на ГТС атом-атомный потенциал Рс up) с ( гтс) может быть перенесен и на адсорбцию на ГТС других алкенов. [27]
Этот потенциал получил широкое применение; так, он является основным в методе атом-атомных потенциалов [13-16], аппроксимирующем потенциал межмо - у лекулярттого взаимодействия суммой атом-атомных взаимодействий. [28]
Таким образом, из рассмотренных выше способов оценки г наиболее надежные значения получаются при использовании атом-атомных потенциалов. Ниже приведены значения s графитирован-ной сажи Р-33 ( Sterling FT, 2700), полученные из приведенных в табл. IX8 наиболее надежных значений szfl [ определенных при использовании потенциалов ( IX2) и ( VIII49) ] и из приведенных выше ( см. стр. [29]
Страницы: 1 2 3 4