Природа - каталитическая активность
Cтраница 3
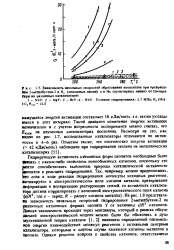 |
Зависимость начальных скоростей образования нзопентана при превращении 2-метилбутена - 2 в Н, ( сплошные линии и в Не ( пунктирные линии от температуры иа различных катализаторах. [31] |
Гидрирующую активность катионных форм цеолитов необходимо было фязать с каким-либо свойством ионообменных катионов, поскольку это Могло способствовать выяснению природы каталитической активности деолитов в реакциях гидрирования. На рис. 1.8 представлена зависимость начальных скоростей гидрирования 2-метилбутена - 2 на различных катионных формах цеолита Y от величины e / R катионов. Данная зависимость проходит через максимум, который в рамках обсуждаемой электростатической модели можно было бы объяснить в духе мультиплетной теории катализа [1,2] наличием определенной оптимальной энергии взаимодействия молекул реагентов с активными центрами катализатора, которыми в данном случае являются катионы металлов в Цеолите. [32]
Однако, прежде чем перейти к рассмотрению изомерных превращений непредельных углеводородов в присутствии алюмосиликатов, необходимо коротко остановиться на природе каталитической активности алюмосиликатного катализатора. [33]
Следует особо отметить своеобразие и сложность изомерных превращений непредельных углеводородов в присутствии алюмосиликатов, что заставило нас в первую очередь осветить ряд вопросов, связанных с природой каталитической активности этого катализатора, а также с химизмом протекающих на нем реакций. [34]
Однако сомнения в ионизации углеводородных молекул в гетерогенных реакциях, протекающих с участием катализаторов кислотного типа ( алюмосиликаты, активированная окись алюминия), не должны касаться природы каталитической активности последних. Наоборот, ряд экспериментальных данных по дезактивации этих катализаторов ионами натрия и органическими основаниями определенно свидетельствуют о присутствии на их поверхности протонизированных атомов водорода. [35]
Гидрообессеривающая активность АКМ и АНМ катализаторов зависит от многих факторов: химического состава и соотношения компонентов, структуры объемных и поверхностных фаз, природы носителя и промотора, условий получения и предварительной обработки, текстуры, размера и формы частиц и др. Выяснению природы каталитической активности такой сложной системы с целью создания физико-химической модели, как основы направленного синтеза катализаторов гидроочистки с заданными свойствами, посвящены многочисленные работы. Однако вопрос пока еще далек от окончательного решения. Тем не менее кажется целесообразным рассмотреть найденные экспериментально корреляционные закономерности влияния отдельных факторов и основные модели катализаторов гидрообес-серивания для того, чтобы наметить пути их совершенствования. [36]
Вкратце проблема сводится к следующему. Природу каталитической активности поверхности связывают с присутствием локальных электронных уровней в запрещенной зоне, между потолком заполненной валентной зоны и зоной проводимости, а также с таммовскими, или локализованными, поверхностными уровнями. Эти электронные уровни возникают в результате наличия дефектов в строении твердого тела, особенно от таких нарушений кристаллической решетки, как дефекты Шоттки и Френкеля, примеси ( которые в катализе могут являться синонимом промотора) и физические нерегулярности типа дислокаций. Эти и другие электронные характеристики подробно рассматриваются во многих книгах по физике твердого состояния [27, 49, 65], а применение этих воззрений к проблемам катализа освещено, например, в публикациях Грея [27, 28], Стоуна [65, 66], Волькен-штейна [81] и других. [37]
Определил природу избыточной каталитической активности, возникающей при ионизирующем облучении окисных катализаторов, и выяснил, какую роль играют в радиационном катализе кислородные дырочные центры. [38]
Исследования катализаторов крекинга разделяются на две основные группы. К первой относится изучение природы каталитической активности алюмосиликатов и механизма крекинга углеводородов различной структуры. Вторая группа охватывает разработку методов лабораторного контроля качества катализаторов при их разработке, производстве и последующем промышленном использовании. При проведении исследований, относящихся к первой группе, в качестве сырья преимущественно применяют индивидуальные углеводороды, а крекинг осуществляют в проточных, про-точно-пульсационных и импульсных установках. В исследованиях, относящихся ко второй группе, используют какое-либо реальное сырье, причем крекинг проводят в проточных условиях. [39]
Однако процессу гидроформинга приходится в настоящее время выдерживать большую конкуренцию со стороны много - численных разновидностей каталитического риформинга в присутствии полифункциональных катализаторов. Механизм реакций углеводородов различных классов, а также природа каталитической активности полифункциональных катализаторов и некоторые вопросы их промышленного использования будут рассмотрены дальше, в соответствующих главах настоящего раздела. [40]
Несмотря на то, что превращения оле-фйновых углеводородов в присутствии активированной окиси алюминия несколько отличны от превращений тех же углеводородов на алюмосиликатных катализаторах, мы считали, что изложение этого вопроса удобнее, в некоторых случаях, вести параллельно, сопоставляя свойства этих катализаторов. В то же время вопросы, связанные с природой каталитической активности окиси алюминия, будут изложены несколько позже - в главе, посвященной реакциям высокомолекулярных углеводородов на. [41]
Действительно ли протон, участвующий в карбоний-ионных реакциях, принадлежит гидроксилу, а не воде, находящейся в каналах цеолита в особом состоянии, отличном от ее обычного и поэтому не проявляющемся спектроскопически. Это соображение не вносит существенных изменений в изложенные выше представления о природе каталитической активности цеолитов, поскольку между различными формами цеолитно-связанной воды может существовать равновесие. Однако оно предполагает, что при известных температурных условиях становится подвижным водород воды, связанной с цео-литным каркасом, а не водород гидроксила. При принятии гидроксильного механизма трудно объяснить сравнительно легкую обратимость эффекта промотирова-ния. В самом деле, если учесть, что число активных центров составляет - 4 % от общего числа обменных мест, то следует принять, что эти активные центры находятся на значительных расстояниях друг от друга. В этом случае, бесспорно, процесс рекомбинации ОН-групп в воду в процессе дегидратации должен быть затруднен. В работе [37] методом ЯМР наряду с протонами, принадлежащими структурным гидроксильным группам, также были обнаружены протоны, принадлежащие адсорбированной воде, хотя после термообработки ( 500 С, 10 - 5 мм рт. ст.) в ИК-спектре образца мы не наблюдали деформационных колебаний молекулярной воды. Наличие особых форм прочно связанной воды в катионных и декатинированных цеолитах представляет несомненный интерес, но авторы не располагают какими-либо данными об участии этой воды в катализе. [42]
Имеющиеся в литературе высказывания по механизму действия подобных катализаторов пока должны рассматриваться лишь как предварительные соображения, не подкрепленные необходимыми экспериментальными фактами. Дальнейшее расширение наших представлений в этой области немыслимо без всестороннего экспериментального изучения природы каталитической активности окисных катализаторов в реакциях полимеризации и механизма самого процесса. Имея в виду сказанное, небезынтересно рассмотреть здесь некоторые экспериментальные наблюдения, касающиеся применения окиснохромовых или окисномолибденовых катализаторов при получении кристаллических поли-а-олефинов, в первую очередь полиэтилена и полипропилена. Как сейчас уже известно, активность окиснохромо-вого и окисномолибденового катализаторов объясняется легкостью перехода металла из одного валентного состояния в другое. [43]
Однако после выполнения исследований с дейтерированными веществами было найдено, что в присутствии промоторов ( НХ) возрастает скорость как изомеризации, так и дейтерообмена. Тогда чтобы объяснить природу каталитической активности соединения НА1Х4, было предположено, что водород от катализатора присоединяется к вторичному углеродному атому в н-парафине, вследствие чего ослабляется и разрывается С - С-связь, Далее катализатор отнимает водород у углеводорода, что приводит к перестройке углеродного скелета последнего. [44]
С одной стороны, исследования в области катализа принесли богатый фактический материал, обобщенный в соответствующие теории. Этот материал позволяет: а) установить природу каталитической активности твердых тел, в том числе стенок, растворителей и малых примесей; б) определить характер участия катализатора в процессе; в) во многих случаях установить механизм реакций, в том числе общие пути снижения энергии активации; г) выявить распространенность определенных типов механизма реакций н общую распространенность катализа в целом; д) в большинстве случаев убедиться в достоверности полученных теоретических выводов путем проверки их в условиях производственной или лабораторной практики. Диалектико-материали-стическая методология, с другой стороны, позволяет лучше ориентироваться во всем этом материале, для того чтобы найти наиболее существенные связи между разными типами катализа, определить общность между ними, выявить причинную обусловленность того факта, что природа столь широко пользуется каталитическим посредничеством в химических превращениях. [45]
Страницы: 1 2 3 4