Проба - анализируемое вещество
Cтраница 1
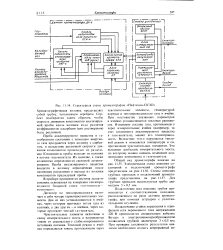 |
Структурная схема хроматографов Нефтехим-СКЭП. [1] |
Проба анализируемого вещества в газообразном состоянии с помощью инертного газа продувается через колонку с сорбентом, и вследствие различной скорости движения компонентов происходит их разделение. Компоненты пробы выходят из колонки в потоке газа-носителя. Их наличие, а также количество определяются системой детектирования. Проба анализируемого вещества вводится в колонку периодически после окончания разделения и выхода из колонки компонентов предыдущей пробы. [2]
Проба анализируемого вещества в паровой фазе вносится в колонку потоком газа-носителя. В процессе его движения вдоль неподвижной фазы-сорбента происходят многократные акты сорбции и десорбции молекул всех компонентов смеси; свойства сорбента подбираются таким образом, чтобы молекулы разных компонентов по разному удерживались им, следовательно, продвигались вдоль колонки с разными скоростями. Поэтому введенная проба по мере движения вдоль колонки постепенно делится на зоны отдельных компонентов, растворенных в газе-носителе. Количество каждого компонента в потоке газа-носителя определяется на выходе из колонки системой детектирования. [3]
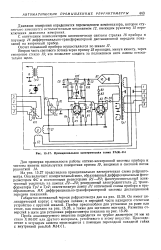 |
Принципиальная электрическая схема РАЖ-451. [4] |
Проба анализируемого вещества подводится по трубке диаметром 14 мм из стали Х18Н9Т или другого материала, устойчивого к этому веществу. [5]
 |
Простейшая камера для круговой тонкослойной хроматографии. [6] |
Пробы анализируемых веществ объемом 0 001 - 0 005 мл ( в случае нанесения в виде точек) или до 0 1 - 0 2 мл ( при нанесении в виде полоски рядом капля за каплей) наносят при помощи пипеток или специально калиброванного стеклянного капилляра. [7]
Проба анализируемого вещества вводится в испаритель с помощью дозирующего устройства. Для ввода газообразных веществ применяют газовые пипетки, жидкости вводят с помощью микрошприца. Пробы твердых веществ вводят в виде раствора в подходящем растворителе. [8]
Проба QafJ анализируемого вещества увлекается потоком инертного вещества-носителя и проходит через слой сорбента, заполняющего хроматографическую колонку. Скорость движения отдельных компонентов анализируемой смеси вдоль колонки зависит от интенсивности процессов сорбции: наибольшую скорость движения имеют компоненты, которые сорбируются слабее других и наоборот. Если условия для проведения анализа выбраны правильно, то за время движения через слой сорбента проба успеет полностью разделиться на отдельные компоненты, которые будут выделяться на выходе хроматографической колонки в определенной последовательности. [9]
Пробу анализируемого вещества нагревают и по уменьшению массы ( косвенный метод) или по массе улавливаемой воды ( прямой метод) можно определить ее содержание. Несмотря на простоту таких методов, при их применении возможны трудности. Прежде всего возникает вопрос, до какой температуры необходимо нагревать пробу. Многие вещества теряют воду при очень высокой температуре: например, кремневая кислота, гидроксид алюминия и другие материалы теряют воду при 1000 С и выше. Кроме того, при нагревании пробы могут происходить и другие процессы. Так, удаляются или разлагаются многие летучие и органические вещества, происходит окисление многих веществ. Улетучивание и разложение соединений будет давать прибавку к массе, которая укажет на присутствие воды, а процессы окисления, наоборот, уменьшают массу, и, таким образом, получится заниженный результат. При сгорании многих органических веществ также может образоваться вода, и поэтому даже при прямом методе ее определения будут получены завышенные результаты. Поэтому гравиметрические методы определения воды весьма приближенны, и при их проведении необходимо придерживаться условий, принятых для определения того или другого вида воды. [10]
Пробу анализируемого вещества ( 0 8 - 2 мг) в кварцевой ампуле вводят в вертикальный реактор, заполненный сажей. При высокой температуре ( обычно выше 1000 С) происходит восстановительная деструкция органического вещества с образованием оксида углерода, количество которого эквивалентно содержанию кислорода в пробе. Конверсия происходит в атмосфере аргона, содержащего 8 % водорода. Затем оксид углерода на слое катализатора конвертируют в метан. Образующийся метан не регистрируется катарометром, так как его теплопроводность практически равна теплопроводности используемого смешанного газа-носителя. Регистрируемый сплав пропорционален вакансии водорода, образующегося в смешанном газе-носителе ( 8 % водорода в аргоне) в результате гидрирования оксида углерода. [11]
Пробу анализируемого вещества подготовляют к определению свинца так, чтобы в растворе было от 1 до 5 мг свинца в виде нитрата и чтобы раствор был свободен от мешающих веществ. К такому раствору прибавляют разбавленную азотную кислоту или разбавленную щелочь, чтобы привести его рН приблизительно к 3 ( капельная проба на индикаторной бумаге) и разбавляют до 50 мл. Затем прибавляют в следующем порядке: 5 мл пиридиновой буферной смеси, 1 мл раствора индикатора и 15 мл перегнанного ацетона, после чего титруют титрованным раствором фосфата, пока розовое окрашивание не превратится в бледножелтое. [12]
Массу пробы анализируемого вещества, отбираемой для количествен - ного определения макрометодом, измеряют долями грамма, микрометодом - - несколькими миллиграммами, а ультрамикрометодом - несколькими микрограммами. Между тем на производстве обычно приходится иметь дело с большими количествами сырья, полупродуктов, готовой продукции и других технических материалов, измеряемыми многими сотнями тонн. Поэтому очень важно, чтобы химический состав отбираемой для анализа пробы в точности соответствовал среднему химическому составу всей партии анализируемого продукта. [13]
Массу пробы анализируемого вещества, отбираемой для количест венного определения макрометодом, измеряют долями грамма, микро методом-несколькими миллиграммами, а ультрамикрометодом-несколь кими микрограммами. Между тем на производстве обычно приходится иметь дело с большими количествами сырья, полупродуктов, готовой продукции и других технических материалов, измеряемыми многими сотнями тонн. Поэтому очень важно, чтобы химический состав отбираемой для анализа пробы в точности соответствовал среднему химическому составу всей партии анализируемого продукта. [14]
Отбор проб анализируемого вещества и титрование необходимо проводить в определенных условиях; в частности, следует пользоваться секундомером, чтобы продолжительность титрования была вполне определенной. Описываемый метод, повидимому, не является вполне удовлетворительным. Уже тот факт, что необходимо тщательно соблюдать и контролировать условия проведения всех операций, свидетельствует либо о том, что экстрагирование воды из образца осуществляется с недостаточной полнотой, либо о том, что при этом методе не удается полностью защитить образец от попадания воды из воздуха. При применении указанного выше растворителя, повидимому, не получается однородного раствора в случае образцов с небольшим содержанием воды. Трудности, связанные с образованием эмульсий, возрастают при применении бензола. Замена последнего декалином приводит к лучшим результатам ( см. стр. [15]
Страницы: 1 2 3 4