Проба - жидкая фаза
Cтраница 4
Если высокая степень точности не обязательна, а давление и температура близки к атмосферным, для измерения растворимости твердых тел в жидкостях пригодны достаточно простые методы и приборы. Две фазы встряхивают вместе до достижения равновесия, после декантации или фильтрования отбирают пробу жидкой фазы, которую далее анализируют химическими методами или проводят ее испарение. Другой широко распространенный метод заключается в постепенном охлаждении горячего концентрированного раствора с регистрацией температуры, при которой происходит осаждение. [46]
Впервые в СССР в 1959 г. А. С. Великовский и В. В. Юшкин предложили определять минимально необходимую скорость ( или дебит скважины) по неизменности фракционного состава жидкого конденсата, поступающего из скважины. Для этого необходимо проводить специальные исследования скважин с различными возрастающими по величине дебитами, отбирать пробы жидкой фазы при работе скважины на установившемся режиме, проводить их разгонку в лабораторных условиях, определяя скорость, при которой и выше которой фракционный состав практически не изменяется. Метод надежен, физически обоснован, однако требует значительных затрат времени и сложного лабораторного оборудования. [47]
Для определения величины D методом изотермической перекристаллизации в сосуд для определения растворимости вводят 25 мл исходного насыщенного раствора NiSCh, содержащего Й9ре2, и 3 5г мелкорастертого перекристаллизованного сернокислого никеля. Смесь помещают в термостат при 25 С, где ее перемешивают винтовой мешалкой, прерывая перемешивание для отбора проб жидкой фазы. [48]
Так как для различных систем необходимо различное время для установления равновесия, то перед изучением каждой новой системы или серии подобных систем определяют время, в течение которого устанавливается равновесие. В связи с тем, что газ насыщается жидкостью быстрее, чем жидкость газом, об установлении равновесия обычно судят по совпадению составов проб жидкой фазы, отбираемых периодически через каждые 15 - 20 мин перемешивания при одних и тех же давлении и температуре. [49]
Изучение фазовых равновесий в системе NaO-NaOH - Н2О методами кривых р-х и р - t оказалось малоэффективным, так как углы пересечения ветвей этих кривых были недостаточно четкими, и потому эта система в основном была изучена методом отбора проб для анализа наряду с параллельным определением давления пара растворов в автоклаве описанного выше устройства. Применение метода отбора проб в данном случае было вполне целесообразно, так как присутствие в растворах некристаллизующейся гидроокиси натрия устраняло возможность забивания фильтра в процессе отбора проб жидкой фазы для анализа. [50]
 |
Структура диаграммы фазового равновесия системы ацетон - вода - толуол - бутилацетат. [51] |
Из рассмотренных вариантов отобраны два варианта технологической схемы разделения. Экспериментально проверена их работоспособность на установке, состоящей из насадочной колонны непрерывного действия диаметром 25 мм и эффективностью - 35 т.т. Колонна была оборудована пробоотборниками для отбора проб жидкой фазы по высоте колонны. [52]
Пробы жидкой фазы при 75 - 150 отбирались после выдерживания в термостате в течение получаса. [53]
Общий метод построения изотермы растворимости заключается в следующем. Положение этого разреза определяется с помощью предварительно поставленных опытов. Смеси приводят в равновесие и отбирают пробы жидкой фазы для анализа. Полученная кривая является изотермой растворимости. [54]
Растворимость в системе СаО - Р2О5 - МН3 - Н20 при 25, 40 и 50 7 С в полях кристаллизации г. о: ю - к дикальцийфосфатов изучали методом изотермической кристаллизации. В процессе изотермической кристаллизации твердых фаз из растворов периодически отбирали пробы жидкой фазы при помощи пипетки, снабженной на конце плотным стеклянным фильтром. [55]
Очистка теплоносителя от загрязняющих его веществ, которые составляют с ним гомогенную систему, является в данном случае наиболее специфической и сложной задачей. В настоящий момент нет возможности представить достаточно полно вид химических соединений радиоактивных элементов, которые при рабочих параметрах газожидкостного цикла реактора составляют гомогенную систему с теплоносителем. В газовой фазе это могут быть соединения йода, элементарный йод, благородные газы, окислы и соединения стронция, бария, хрома, молибдена, цезия, углерода и рутения. В пробах жидкой фазы теплоносителя гамма-спектрофотометрическим методом обнаружены незначительные количества железа, кобальта и рутения. Происхождение последних может быть обусловлено двумя причинами: высокодисперсным состоянием твердой фазы соединений этих элементов и наличием соответствующих растворимых в N2O4 соединений. [56]
Раствор фосфорной кислоты, содержащий NH, заливали в термостатированную четырехгорлую колбу, куда загружали заведомый избыток сульфата кальция. Во время перемешивания периодически отбирали пробы жидкой фазы с помощью пипетки с фильтром Шотта № 2 или № 3 для определения содержания окиси кальция. [57]
Предварительно были построены соответствующие зависимости показателей преломления жидкой фазы. Для определения показателя преломления периодически отбирались пробы жидкой фазы. [58]
В аналитических или препаративных колонках, работающих в нормальных условиях, не происходит сколько-нибудь значительного перераспределения содержания жидкой фазы на носителе. Такое перераспределение происходит обычно в колонках, работающих с перегрузкой, а поскольку с перегрузкой работает большинство препаративных колонок, то это явление происходит гораздо чаще, чем кажется. Вследствие малой эффективности многих препаративных колонок отделение жидкой фазы от носителя часто остается незамеченным. При введении в колонку проб больших величин, превышающих емкость колонки, концентрация компонентов разделяемой смеси в жидкой фазе оказывается очень высокой. При этом происходит не только растворение компонентов разделяемой смеси в жидкой фазе, но и заметное растворение в пробе жидкой фазы. По мере прохождения компонентов смеси через колонку происходит расширение хроматографической полосы и уменьшение локальной концентрации компонентов. Вследствие этого уменьшается концентрация жидкой фазы, растворенной в разделяемой пробе, и она вновь осаждается на носителе. Весь этот процесс происходит на начальном участке колонки длиной около метра. В результате носитель в начальном участке колонки остается без жидкой фазы, а за этим участком образуется область с повышенной концентрацией жидкой фазы. Такое локальное повышение концентрации жидкой фазы приводит к расширению хроматографической полосы вследствие увеличения времени пребывания разделяемой смеси и уменьшению эффективности колонки. Носитель на начальном участке колонки, лишенный жидкой фазы, действует как перколяционный фильтр, через который проходит разделяемая смесь прежде, чем она попадет в область с носителем, покрытым жидкой фазой, где и происходит ее разделение. [59]
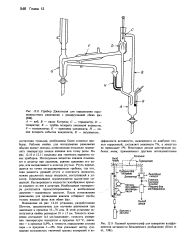 |
Прибор Джиллеспи для определения паро-жидкостного равновесия с рециркуляцией обеих фаз.| Газовый хроматограф для измерения коэффициентов активности бесконечного разбавления ( Diner et al., 1981. [60] |
Страницы: 1 2 3 4 5