Кинетическая величина
Cтраница 1
Экстраполяция кинетических величин на более низкие температуры неизбежно связана с внесением погрешности. Если аррениусовский ход параметра действительно сохраняется при более низкой температуре, то главный источник погрешности - неточность в измерении энергии активации. [1]
Для кинетических величин разнообразие возможных случаев возрастает в связи с возможным разнообразием форм уравнений движения, описывающих релаксацию. Обсудим сначала простейший случай однокомпонентного параметра порядка ( B. [2]
Как выражать кинетические величины через атомные константы, из таких формул не следует. [3]
Если обе кинетические величины, kRC и & д о, могут быть получены на основе экспериментальных измерений, то можно определить сумму подвижностей. [4]
Указанная схема расчета кинетических величин условно была применена для построения кинетических зависимостей и для других карбидно-окисных композиций в соответствии с наиболее вероятными ( по данным термодинамического анализа) уравнениями реакций. [5]
Существенно также, что кинетические величины, рекомендуемые Крайнем и Смитом, более или менее точно характеризуют процесс на американском катализаторе и едва ли могут быть использованы для расчетов реакций на отечественном катализаторе. Вместе с тем работа Смита создает предпосылки для создания точной модели и исполь-г зевалась в качестве исходной многими авторами. [6]
Такая теория должна связать макроскопические кинетические величины с новыми величинами, используемыми для описания молекул. Хотя решение такой задачи в принципе возможно, но оно слишком трудно. Поэтому целесообразно выбрать менее полную систему молекулярных единиц, такую, чтобы она давала возможность связать макроскопические величины с важнейшими параметрами молекул. Иными словами, следует избрать некоторую целесообразную модель молекулы, достаточно простую для математического расчета и такую, чтобы ее свойства можно было связать с другими экспериментально определенными свойствами молекул. В следующей главе мы познакомимся с некоторыми из таких общепринятых моделей и рассмотрим математический аппарат для их описания. [7]
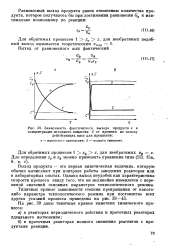 |
Зависимость фактического выхода продукта х и концентрации исходного вещества С от времени по закону. [8] |
Выход продукта - это первая кинетическая величина, которую обычно вычисляют при контроле работы заводских реакторов или в лабораторных опытах. Однако выход неудобен как характеристика скорости процесса ввиду того, что он нелинейно изменяется с переменой значений основных параметров технологического режима. [9]
Таким образом, определение кинетических величин и их дисперсий в рассматриваемых реакторах не вызывает затруднений. [10]
 |
Величины энергий активации первой стадии реакции.| Определение кинетических величин второй стадии реакции каталитического крекинга тяжелого нефтяного газойля. [11] |
Значительный интерес представляет определение кинетических величин при каталитическом крекинге не только для первой, но и для второй стадии реакции, протекающей по схеме ( VI-6), причем А - сырье, В - жидкие продукты реакции, С - кокс, D - газ. [12]
Уравнение Флори не требует знания кинетических величин, однако оно связано с ними. [13]
Можно добиться прецизионной точности измерения кинетических величин, но какое отношение имеют эти оценки непосредственно к динамике электронного облака. [14]
В задачах НРГД необходимо описание дополнительных кинетических величин - источников и стоков спектральной интенсивности излучения, связанных с неравновесностью распределения ионного компонента плазмы по внутренним степеням свободы. В стандартных моделях НРГД в области высоких температур, при которых степень ионизации плазмы достаточно велика, эти величины описываются в рамках радиационно-столкно-вительных моделей ( РСМ) кинетики плазмы для концентраций ионов и их возбужденных состояний. [15]
Страницы: 1 2 3 4