Растворение - проба
Cтраница 1
Растворение пробы проводят в вытяжном шкафу сначала без нагревания под часовым стеклом. По окончании бурной реакции колбу нагревают на песчаной бане до слабого кипения до полного растворения навески и удаления оксидов азота. На дне колбы может остаться графит черного цвета, который при взбалтывании плавает. Добавляют в колбу 60 - 70 мл горячей воды, 5 мл 0 5 % - ного раствора нитрата серебра и 10 - 15 мл 25 % - ного раствора персульфата аммония. Раствор кипятят 1 5 - 2 мин, но не более. За начало кипения принимают момент образования первых крупных пузырьков по всему объему раствора. Раствор приобретает фиолетово-красную окраску и должен оставаться прозрачным. Если образовался бурый осадок оксида дигидроксида марганца МпО ( ОН) 2, то зто значит, что НМпО4 частично разложилась; появление белой мути AgCl ходу анализа не мешает. Через 2 - 3 мин колбу охлаждают под краном, закрыв горловину колбы стаканом. [1]
Растворение пробы проводят при нагревании в течение часа. Параллельно ставят три опыта. Затем растворы переносят в мерные колбы емкостью 25 мл, нейтрализуют H2SO4 и доводят объем до метки водой. Растворы перемешивают, заливают в кюветы емкостью 5 мл и включают секундомер. Оптическую плотность растворов измеряют через 30 мин. [2]
Растворение пробы и отделение сурьмы производят так же, как описано в гл. NaOH, 15 капель пергид-рола и кипятят 5 мин. Полученный щелочной раствор переносят в коническую колбу, фольгу и стакан ополаскивают два-три раза возможно малыми порциями воды, осторожно добавляют в колбу 1 5 мл H2SO4 ( уд. Содержимое колбы нагревают 10 мин. [3]
Растворение пробы по возможности осуществляют действием одной азотной кислоты, так как окисление селена и теллура происходит только до четырехвалентного состояния и нет опасности потерь селена. Если применяется царская водка или соляная кислота, надежнее пользоваться колбой с обратным холодильником. Нерастворимые в кислотах минералы сплавляют с перекисью натрия. Для глинистых л кремнистых руд полезно разложение по Кьельдалю ( см. разд. [4]
Растворение пробы проводят при нагревании в течение часа. Параллельно ставят три опыта. Затем растворы переносят в мерные колбы емкостью 25 мл, нейтрализуют H2SO4 и доводят объем до метки водой. Растворы перемешивают, заливают в кюветы емкостью 5 мл и включают секундомер. Оптическую плотность растворов измеряют через 30 мин. [5]
Растворение проб в кислотах или щелочах следует проводить только в вытяжном шкафу. [6]
После растворения пробы в 5 - 10 % - ном растворе H2SO4 для отделения от посторонних элементов повторяется, по существу, метод фторидного вскрытия. [7]
Для растворения проб, подкисления или иодщелачивания, осаждения и многих других операций к исследуемому объекту добавляют очень малые объемы растворов реактивов, так как при уменьшении объемов анализируемых растворов соответственно уменьшаются и объемы вводимых растворов реактивов. Следовательно, относительное влияние загрязнений в реактивах одинаково как в макро -, так и в ультрамикроанализе. [8]
После растворения пробы раствор разбавляют до определенного объема в мерной колбе ( еще лучше в ней же провести и растворение) и к его аликвотной части прибавляют серную кислоту и раствор молибдата аммония. Раствору дают постоять несколько минут для образования гетерополикислоты, а затем к нему прибавляют H2SO4 и раствор SnCl2 в качестве восстановителя. Полученный раствор разбавляют до определенного объема и фотомет-рируют. [9]
Для растворения пробы авторы рекомендуют смесь этиленгликоля с пиридином. Благодаря отсутствию метилового спирта мешающие влияния сводятся к минимуму или вообще устраняются. Стабилизованный реактив изготовляется промышленностью и поступает в продажу. [10]
После растворения пробы в азотной кислоте добавляют небольшой объем концентрированной серной кислоты, и раствор кипятят для разложения нитратов и удаления оксидов азота, которые способны окислять иодид. Если сернокислый раствор разбавить водой и охладить, то сульфат свинца почти количественно осадится и должен быть удален фильтрованием. В противном случае, после добавления иодида для определения меди образуется светло-желтый иодид свинца ( РЫ2), который не позволяет четко определить конечную точку титрования. [11]
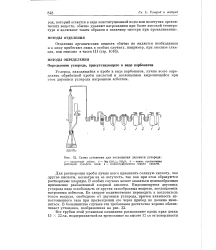 |
Схема установки для поглощения двуокиси углерода. [12] |
Для растворения пробы лучше всего применять соляную кислоту, чем другие кислоты, несмотря на ее летучесть, так как при этом образуются растворимые хлориды. В особых случаях может оказаться целесообразным применение разбавленной хлорной кислоты. [13]
Для растворения проб используют перегнанную азотную кислоту и дважды дистиллированную воду. Перегонка кислоты и воды производится в приборах, изготовленных из прозрачного кварца. Чтобы проконтролировать отсутствие случайных загрязнений при растворении и усреднить результаты, параллельно растворяют две навески. [14]
После растворения пробы прибавляют около 50 мг сульфамида и выпариванием удаляют окислы азота. Охлаждают, добавляют 25 мл 8 % - ного раствора йодида КРЛИЯ и выделившийся свободный йод оттитро-вываюг тиосульфатом в присутствии крахмала в качестве индикатора. Таким способом определяют медь, если количеством присутствующего в растворе железа можно пренебречь. Если желательно определить медь и железо, поступают несколько измененным способом. Навеску сплава растворяют в азотной кислоте, не добавляя винной кислоты. Нейтрализуют концентрированным раствором аммиака ( около 6 мл) и добавляют столько твердого фторида калия ( 0 2 - 2 0 г), чтобы выпавший осадок снова растворился. После этого йодометрически титруют медь. [15]
Страницы: 1 2 3 4