Расчет - поверхность - потенциальная энергия
Cтраница 2
Эти методы интенсивно используются для определе-ния равновесных конфигураций молекул, а также для расчета поверхностей потенциальной энергии молекулярных систем при анализе механизма хим. р-ций. [16]
Ситуация здесь в общем случае та же, что и в теории абсолютных скоростей реакций, которая содержит расчет поверхностей потенциальной энергии для реагирующей системы и устанавливает связь фактических скоростей реакций с этими потенциальными поверхностями. Эта связь исследовалась Эйрингом и др. [9] на основе допущения равновесия между реагентами и активированными комплексами, которые существуют в окрестности седловой точки поверхности потенциальной энергии. Такое предположение о равновесии строго обосновать нельзя, и для получения более точных сведений о взаимодействии реагирующих частиц нужно решить их уравнения движения при наличии связей в виде поверхностей потенциальной энергии. [17]
 |
Путь реакции Y XR - - - YX R ( более детальное изображение профиля поверхности потенциальной энергии реакции на. [18] |
Для выяснения механизма реакций Х - перехода наиболее существенны два свойства активированного комплекса: его положение на пути реакции, в частности, расположен ли он в районах А, В или С ( см. рис. 15.1), и высота энергетического барьера. Расчет поверхности потенциальной энергии позволяет естественно связать оба эти свойства активированного комплекса. [19]
Ни один из этих методов не может быть применен для всего широкого диапазона, в котором изменяется реакционная способность и селективность в вышеупомянутых типах химических реакций. Квантовомеханические методы расчета поверхностей потенциальной энергии реакций все еще слишком грубы для учета тонких различий в скоростях или направлениях ориентации в зависимости от небольших изменений в заместителе. В любом случае получение всех точек, необходимых для точного определения изменения энергии при изменениях углов и длин связей в трехмерном пространстве, является очень трудоемким и дорогостоящим занятием. Тем не менее тонкие детали механизмов реакций, касающиеся синхронности и природы элементарных стадий, не могут быть получены никаким другим путем. Например, недавние расчеты энергетических поверхностей для циклофрагментации циклобутана с образованием этилена [31] и для геометрической изомеризации циклопропана [32] показали, что промежуточным бирадикальным частицам ( например, тетраметилен и триметилен) не соответствуют минимумы на поверхности потенциальных энергий этих реакций. [20]
 |
Реакции о-замещенных бензилхлоридов. [21] |
Явление нуклеофильной реакционной способности очень сложно, и окончательный анализ требует расчетов энергий активации по методу ab initio, включая участие растворителя. Несмотря на значительный прогресс в области расчета поверхностей потенциальной энергии органических реакций, происшедший в последние несколько лет и стимулированный детальными исследованиями электроциклических реакций, теоретическое рассмотрение гетеролити-ческих процессов в растворе представляет исключительно трудную задачу. [22]
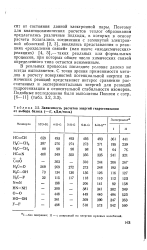 |
Зависимость расчетов энергий гидрогенизации от выбора базиса ( - Е, кДж / моль. [23] |
В реальных процессах последнее условие далеко не всегда выполняется. С точки зрения применимости методов к расчету поверхностей потенциальной энергии химических реакций представляет интерес сравнение рассчитанных и экспериментальных энергий для реакций гидрогенизации и относительной стабильности изомеров. [24]
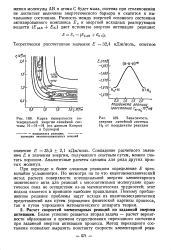 |
Карта поверхности потенциальной энергии линейной системы Н - Н - Н ( по данным Конроя и Б рун ер а.| Зависимость энергии линейной системы Н3 от координаты реакции. [25] |
При переходе к более сложным реакциям определение Е чрезвычайно усложняется. Но несмотря на то что квантовомеханический метод расчета поверхности потенциальной энергии элементарного химического акта для реакций с многоатомными молекулами практически пока не осуществим из-за математических трудностей; этот подход является в принципе наиболее правильным. [26]
Реагирующая система ( квадрициклан - норборнадиен) описана в простой четнрвзсэлектроЕяой модели с учетом взаимодействия электронов. Перезаселэние граничных орбиталей разной симметрии, характерное для реакций с запретом по орбитальной симметрии, учтено в расчете поверхностей потенциальной энергии включением однократно - и двукратно-возбужденных электронных конфигураций. Оказалось, что смешивание возбужденных состояний реагентов с основным состоянием реагентов происходит только в присутствии металлокомплекса. В результате энергия активации каталитического процесса оказывается значительно меньше, чем энергия активации некаталитического процесса. Объяснено влияние на катализ таких факторов, как резонансное взаимодействие реагентов с катализатором, эффекты электронной корреляции в катализаторе и реагирующей системе. [27]
 |
Изменения энергии для различных углов атаки. H-I-Hj ( e. Cl - f - HI ( б. [28] |
Некоторые результаты исследований с помощью молекулярных пучков. Здесь мы ответим на вопросы, поставленные в предыдущем разделе, и покажем, как изучение столкновений и расчет поверхностей потенциальных энергий могут пролить свет на ход реакций. [29]
Однако дальнейшее развитие изучения элементарных актов химических превращений было затруднено отсутствием общей теории для скорости любой реакции в любой фазе, для которой медленным процессом является переход через потенциальный барьер [ 33, стр. Теория абсолютных скоростей реакций, предложенная в 1935 г., опиралась при определении скоростей взаимодействий на методы квантовой ( расчет поверхности потенциальной энергии системы) и статистической ( вычисление абсолютной скорости реакции) механики. [30]
Страницы: 1 2 3