Квантовомеханический расчет
Cтраница 1
Квантовомеханические расчеты так называемого коэффициента свободно-свободного поглощения с различной степенью приближения были проведены рядом авторов. [1]
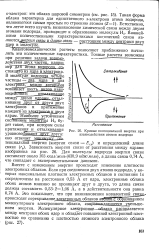 |
Кривые потенциальной энергии при взаимодействии атомов водорода. [2] |
Квантовомеханические расчеты позволяют приближенно вычислить эти количественные характеристики. Точные расчеты возможны при решении задачи взаимо действия двух частиц, напри мер дляатома водошда, со-стояшдо Жядг а иэлектрона. [3]
Квантовомеханические расчеты показывают, что свободный радикал Y, образующийся в результате атаки в - положение, всегда менее устойчив, чем два других, независимо от природы заместителя Z. Из этих же расчетов вытекает, что незамещенное бензольное ядро должно быть менее активным по отношению к радикальным реагентам по сравнению с ядром, содержащим какой бы то ни было заместитель. [4]
Квантовомеханические расчеты показывают, что свободный радикал V, образующийся в результате атаки в м-положение, всегда менее устойчив, чем два других, независимо от природы заместителя Z. Это и является причиной низких выходов jn - замещенных продуктов. Из этих же расчетов вытекает, что незаме щенное бензольное ядро должнб быть менее активным по отношению к радикаль ным реагентам по сравнению с ядром, содержащим какой бы то ни было заместитель. [5]
Квантовомеханический расчет показывает, что по сравнению с катионом ( С5Н) и свободным радикалом ( C5Hs) анион ( С5Н Р) имеет наибольшую энергию сопряжения и, следовательно, стабильнее крайне неустойчивого катиона и свободного радикала. [6]
Квантовомеханические расчеты показывают, что свободный радикал V, образующийся в результате атаки в ж-положение, всегда менее устойчив, чем два других, независимо от природы заместителя Z. Это и является причиной низких выходов ж-замещенных продуктов. [7]
Квантовомеханические расчеты показывают, что при присоединении двух и более электронов к атому энергия отталкивания всегда больше энергии притяжения - сродство атома к двум и более электронам всегда отрицательно. Как мы увидим в дальнейшем ( см. стр. [8]
Квантовомеханические расчеты, проведенные недавно, показали, что вероятность изменения мультиплетности системы при обменном взаимодействии спина с я-связью в комплексе возрастает на много порядков. С позиций локальной активации могут быть рассмотрены химические особенности полисопряженных систем. Увеличение длины системы сопряжения сопровождается повышением подвижности я-электронов и склонности молекул рассматриваемых веществ ко всякого рода перегруппировкам связей и атомов. Перегруппировкой связей с образованием азоформы, протекающей при сравнительно низкой температуре, обусловлен взрывообразный характер процесса деструкции некоторых полимеров. В частности подобные явления характерны для полиазинов, по-видимому претерпевающих при нагревании азоазинную перегруппировку. [9]
Квантовомеханический расчет показывает, что наряду с электростатическим вкладом следует учитывать вклад в энергию водородной связи, определяемый делокализацией электронов. [10]
Квантовомеханический расчет показывает, что наибольшая плотность неспаренного электрона сосредоточена на атомах азота. Значительно меньше локализован неспаренный электрон на а-уг-леродных атомах и еще меньше на р-углеродных атомах. [11]
Квантовомеханический расчет потерь энергии был произведен Бете и Левингстоном. [12]
Квантовомеханические расчеты показывают, что энергия межмолекулярного взаимодействия в случае дальнодействующих сил складывается из энергии возмущения первого порядка - электростатической, и второго порядка - индукционной и дисперсионной. [13]
Квантовомеханические расчеты позволяют вычислить порядок связи, индексы свободной валентности и некоторые другие параметры полимеров, содержащих систему сопряженных связей. Все эти характеристики, отражающие распределение электронной плотности по химическим связям, в ряде случаев позволяют сделать полуколичественные заключения о реакционной способности, структуре переходных комплексов и других свойствах соединений с сопряженными связями. [14]
Квантовомеханические расчеты показывают [23], что по мере роста длины сопряжения энергии связывающей и разрыхляющей орбит сближаются, и при определенной длине имеет место вырождение основного синглетного и триплетного ( разрыхляющего) состояний. Напомним также, что по мере увеличения длины участка сопряжения возрастает и склонность полиеновых структур к цис, трамс-изомеризации. Наглядным подтверждением этого является склонность к подобной изомеризации в ряду каротиноидов. [15]
Страницы: 1 2 3 4