Неэмпирический расчет
Cтраница 1
 |
Расчеты структуры и энергии циклобутадиена в основных триплетном и синглетном состояниях3. [1] |
Неэмпирические расчеты на уровне ССП предсказывают, независимо от базиса, что низшее сингл етное состояние Ai имеет минимум для прямоугольной структуры [11, 15, 16], но без учета корреляционных эффектов квадратное триплетное состояние 3Л2 лежит ниже на 25 кДж / моль. [2]
Неэмпирический расчет в базисе 4 - 31 - G предсказывает для этого катиона плоскую структуру симметрии D3h с длиной связи 0 1076 нм, переход к большему базису 6 - 31 - G дает длину связи 0 1078 нм. [3]
Неэмпирические расчеты по методу валентных схем сравнительно трудно проводить в силу нескольких причин. Простейшие волновые функции метода валентных схем для молекул, имеющих ковалентные связи, всегда состоят из более чем одной детерминантной функции. Вторая причина состоит в том, что проведение алгебраических выкладок для гамильтониановских интегралов и интегралов перекрывания с детерминантными функциями метода валентных схем труднее, чем с детерминантными функциями метода молекулярных орбиталей, так как одно-электронные функции [ tyr ( i) в формуле ( 8 9) ] метода молекулярных орбиталей взаимно ортогональны, в то время как в методе валентных схем они неортогональны. Проведенная оценка показывает, что метод валентных схем был применен менее чем в 5 % неэмпирических расчетов, опубликованных за прошедшие 20 лет, причем ни один из этих расчетов не был посвящен рассмотрению больших молекул. [4]
Неэмпирические расчеты по методу валентных схем сравнительно трудно проводить в силу нескольких причин. Простейшие волновые функции метода валентных схем для молекул, имеющих ковалентные связи, всегда состоят из более чем одной детерминантной функции. Вторая причина состоит в том, что проведение алгебраических выкладок для гамильтониановских интегралов и интегралов перекрывания с детерминантными функциями метода валентных схем труднее, чем с детерминантными функциями метода молекулярных орбиталеи, так как одно-электронные функции [ t v ( 0 в формуле (8.9) ] метода молекулярных орбиталеи взаимно ортогональны, в то время как в методе валентных схем они неортогональны. Проведенная оценка показывает, что метод валентных схем был применен менее чем в 5 % неэмпирических расчетов, опубликованных за прошедшие 20 лет, причем ни один из этих расчетов не был посвящен рассмотрению больших молекул. [5]
Неэмпирические расчеты даже в случае молекул остаются до настоящего времени дорогостоящими и весьма трудоемкими, а их значение определяется главным образом тем, что они имеют контрольный характер по отношению к полуэмпирическим методам. Тем более ясно, что для таких сложных систем, как кристалл с ЛЦ, расчеты электронной структуры практически осуществимы лишь на основе полуэмпирических методов теории молекул. [6]

Неэмпирический расчет у - А12О3 зонным методом ОЛКАО выполнен в работе [46], где проведено рассмотрение зависимости электронных свойств у-сксида от типа координации VAl. Моделировались кубические у-фазы, содержащие вакансии в ( 1) - тетра - и ( 2) - октаэдрических узлах катионной подрешетки, соответствующие ячейки имели составы А15ГУ3ГА1160О32 и А18гА1130Уз0Оз2; здесь AF, V7 и А1, Vю - катион ( вакансия) в тетра - и октапозициях, соответственно. [8]
Неэмпирические расчеты для простых углеводородов в рамках метода Харти-Фока позволяют получить барьеры, весьма близкие к экспериментальным, несмотря на TOV что отличие рассчитанной полной энергии от истинной превышает величину барьера примерно на два порядка. Питцер и Липскомб [90], использовав обычные слетеровские функции и вычислив более 1000 интегралов, получили барьер в этане 3 3 ккал / моль. Позднее Гудисмен [91] указал, что энергии заслоненной и скрещенной форм превышали в этом расчете истинные на 500 ккал / моль, что в 150 раз больше высоты барьера. [9]
Неэмпирические расчеты в приближении сильной связи настолько сложны и сталкиваются с такими вычислительными трудностями, что выполняются очень редко, и, за исключением модельных расчетов одномерных систем [4, 28], пока еще не проводилось систематических исследований проводящих молекулярных комплексов. Трехмерные расчеты в полном приближении для этого класса комплексов с переносом заряда или с водородной связью пока что тоже неосуществимы. По-видимому, это подтверждается тем фактом, что порог электронного поглощения не согласуется с соответствующей термической энергией активации для проводимости. Результаты расчетов методами ЧПДП ( INDO) и МЧПДП ( MINDO) для полиглицина [6], где запрещенные зоны имеют ширину порядка 8 51 - 9 53 эВ, исключают возможность беспримесной проводимости и для этой системы. В работе [147] было установлено, что для электрической проводимости направление полипептидного остова более благоприятно, чем водородные связи. Электронное строение имидазола [18] и полиглицина [39] совершенно явно свидетельствует об образовании в этих системах водородной связи. [10]
Неэмпирические расчеты свойств атомов и молекул уже давно считаются одной из центральных проблем химии, а также атомной и молекулярной физики. Теоретические исследования позволяют устанавливать свойства частиц, которые трудно, а иногда и невозможно изучать экспериментально; это делается путем приближенного решения соответствующих квантовомеханических уравнений. Многие соединения обладают слишком большой реакционной способностью, и вследствие этого их нельзя изолировать и изучать обычными экспериментальными методами, такими, как, например, рентгеноструктурный анализ или инфракрасная спектроскопия. Ди-польный момент заряженных частиц нельзя измерить экспериментально, а для свободных радикалов это удается сделать лишь с большим трудом. Однако такие молекулярные свойства часто поддаются довольно точному неэмпирическому вычислению. Эти частицы обладают очень малым временем жизни в земных лабораторных условиях, однако в космическом пространстве благодаря редким столкновениям их время жизни намного больше. Некоторые химические реакции характеризуются временем протекания порядка пикосекунд, и по экспериментальным данным можно судить об их переходных состояниях лишь косвенно, например по влиянию заместителей на скорость реакции. Теоретические исследования в таких случаях могут дать важные сведения о переходных состояниях. [11]
Однако недавние неэмпирические расчеты показали, что в этом анионе я-электроны почти равномерно распределены по всем пяти атомам [ 27с ], так что в расчетах по простому методу МО для атомов углерода и азота должны быть приняты одинаковые значения кулоновских интегралов. [12]
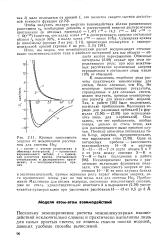 |
Кривые зависимости энергии от межатомного расстояния для системы Не2. [13] |
Поскольку неэмпирические расчеты межмолекулярных взаимодействий исключительно сложны и практически выполнимы лишь для самых простых систем, не лишены смысла поиски моделей, дающих удобные способы вычислений. [14]
Однако точные неэмпирические расчеты координационных соединений все еще остаются очень трудоемкой и весьма дорогостоящей процедурой. [15]
Страницы: 1 2 3 4