Неэмпирический расчет
Cтраница 2
Для неэмпирических расчетов члены R не обяза тельно должны быть полностью пренебрежимыми величинами. [16]
Большинство неэмпирических расчетов в настоящее время сделано в приближении ССП. По крайней мере для небольших молекул используемый базисный набор АО достаточно велик, чтобы приблизиться к хартри-фоковскому пределу. Однако в методе Хартри-Фока рассматривается только одна конфигурация, и поэтому имеется большая ошибка в межэлектронном отталкивании, называемая корреляционной ошибкой. При рассмотрении химической реакции можно было бы надеяться, что корреляционная ошибка будет взаимно уничтожаться для реагентов и продуктов. Факты свидетельствуют о том, что это происходит не часто. [17]
Из неэмпирических расчетов следует, что между е и 2а / ( низшее подходящее возбуждение) существует большой энергетический промежуток. Молекула ВН3 должна быть очень устойчивой по отношению к диссоциации. Молекула BF3 имеет несколько несвязывающих МО, локализованных на атомах фтора ( гл. [18]
Трудности неэмпирических расчетов породили еще более простые ( чем методы НДП) чисто полуэмпирические методы расчета, из которых наибольшее распространение в применении к координационным соединениям получил метод МВГ. Первоначальная идея, предложенная Вольфсбергом и Гельмгольцем [138], сводилась к тому, чтобы в секулярном уравнении метода МО ЛКАО ( V. [19]
Трудности неэмпирических расчетов привели к развитию полуэмпирических методов МО ЛКАО, в которых наиболее сложные для вычислений интегралы в матричных элементах аппроксимируются известными из опыта данными. [20]
Результатам неэмпирических расчетов не всегда удается придать простой химический смысл и простую физическую интерпретацию. Поэтому широко используются полуэмпирические методы МО ЛКАО, в которых молекулярные интегралы не рассчитывают, а вводят как параметры. Кроме того, схему расчета значительно упрощают за счет ряда предположений. [21]
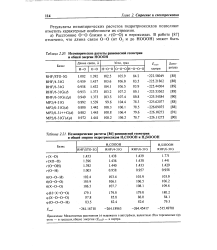 |
Неэмпирические расчеты равновесной геометрии и общей энергии НОООН.| Неэмпирические расчеты равновесной геометрии. [22] |
Результаты неэмпирических расчетов гидротриоксидов позволяют отметить характерные особенности их строения. [23]
При неэмпирическом расчете квантовохимическими методами и при использовании полуклассических методов типа RKR ( Ридберга - Клейна - Риса) и его модификаций потенциал взаимодействия ядер в молекулах находится в виде поточечно заданной функции. [24]
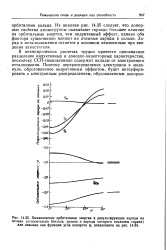 |
Хюккелевские орбитальные энергии и результирующие заряды на. [25] |
В неэмпирических расчетах трудно провести однозначное разделение индуктивных и донорно-акцепторных характеристик, поскольку ССП-гамильтониан содержит вклады от электронного отталкивания. [26]
В неэмпирических расчетах молекулярные орбитали рассматриваются как ЛКАО. Вообще, чем больше набор орбиталей и чем более они подвижны, тем меньше энергия. [27]
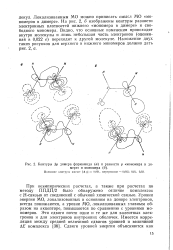 |
Контуры Др дилера формамида ( а и разности р мономера в димере и мономера ( б. [28] |
При неэмпирических расчетах, а также при расчетах по методу ППДП / 2 было обнаружено отличие комплексов с Н - связью от соединений с обычной химической связью. Уровни энергии МО, локализованных в основном на доноре электронов, всегда понижаются, а уровни МО, локализованных главным образом на акцепторе, повышаются по сравнению с уровнями мономеров. [29]
В неэмпирических расчетах методом ССП МО ЛКАО, которые мы рассмотрим сначала, имеется два источника ошибок: 1) однодетерминантное приближение и связанное с этим пренебрежение энергией корреляции и 2) ограниченность базисного набора функций ( см. гл. Ошибки, обусловленные однодетерминантным приближением, в меньшей степени сказываются на расчетах геометрии. Это объясняется тем, что силы, действующие на ядра, являются одноэлектронным свойством и определяются распределением электронной плотности системы. Последнее хорошо передается уже к рамках хартри-фоковского приближения. Это в свою очередь может приводить к трудностям при расчете геометрии подобных систем в недостаточно гибком базисе. [30]
Страницы: 1 2 3 4