Квантовохимический расчет
Cтраница 1
Квантовохимические расчеты методом молекулярных орбиталей главным образом были применены для расчета следующих величин. [1]
Квантовохимические расчеты также применяются для - определения геометрии и других свойств молекул в возбужденном состоянии. Можно указать, например, что, как нашел Хофман ( 1970), метан в одном из возбужденных состояний имеет плоское строение. Но иногда для суждения о возбужденных состояниях достаточно и качественного рассмотрения с позиций квантовой химии. Примером могут служить известные диаграммы Уолша ( 1953), которые применимы к молекулам с небольшим числом атомов в, следовательно, только к простейшим органическим соединениям. [2]
Квантовохимические расчеты также применяются для определе-лия геометрии и других свойств молекул в возбужденном состоянии. Можно указать, например, что, как нашел Хофман ( 1970), метан в одном из возбужденных состояний имеет плоское строение. Но иногда для суждения о возбужденных состояниях достаточно и качественного рассмотрения с позиций квантовой химии. Примером могут служить известные диаграммы Уолша. [3]
Квантовохимические расчеты по расширенному методу Хюккеля позволяют оценить энергию активации каждого из направлений миграции. [4]
Квантовохимические расчеты и данные экспериментов, в которых триметиленме-тильный бирадикал был детектирован, указывают на то, что триметиленметильный би-радикал имеет основное триплетное состояние. В обзоре обсуждаются различные методы генерации указанного бирадикала, его строение и реакции. Исследование термических превращений метиленциклопропана и его производных показало, что образующаяся в промежуточной стадии частица отличается по свойствам от свободного бирадикала. [5]
Квантовохимические расчеты по Хюккелю [19] с использо ванием различных моделей г участием Зй-орбиталей фосфора показали, что только обычная модель Фукуи с / 7я - орбиталью и одним / 7 -электроном и модель Дьюара с двумя ортогональными З - орбиталями гетероатома согласуются с экспериментальными данными по электронным спектрам, реакционной способности и основности 1 1-дифенилфосфорина. [6]
Квантовохимические расчеты были использованы также для оценки реакционной способности [178-184] и интерпретации электронных спектров [185-189] изомерных тиенотиофенов. [7]
Квантовохимические расчеты [56] также пЬдтвердили, что первые волны в случае о - и n - гидроксибензилиденанилинов соответствуют хиноидной форме. [8]
Квантовохимические расчеты позволили установить, что наибольшее влияние на степень защиты ингибитора оказывают дипольный момент, количество атомов в молекуле и количество валентных электронов. [9]
Квантовохимический расчет [25] указывает на ослабление в этом случае стерических препятствий по сравнению со случаем циклодимеризации двух молекул этилена, и тогда процесс, разрешенный по симметрии, осуществляется. [10]
Квантовохимические расчеты проделаны по методу МО ЛКАОнаЭВМ БЭСМ-4М по стандартной программе. [11]
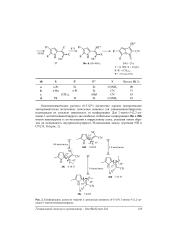 |
Конформации, разности энергий и дипольные моменты ( 6 - 31G 2-метил - 5 - ( 2 2-ди-циано - 1 -метилтио винилпиррола. [12] |
Квантовохимические расчеты ( 6 - 31G) достаточно хорошо воспроизводят экспериментально полученные дипольные моменты для дициановинилпирролов, подтверждая их сильную зависимость от конформации. [13]
Квантовохимические расчеты [47] позволили согласовать дан ные по темновой ( каталитической) и фотохимической димеризаци ] с энергетическими характеристиками переходного состояния. [14]
Квантовохимические расчеты ППЭ представляют очень трудную теоретическую задачу. Этому много причин, мы отметим только некоторые из них. Во-первых, в силу многомерного характера ППЭ для их достаточно полного описания требуется провести квантовохимиче-ский расчет для астрономически большого числа точек дтж. N - число точек по каждой независимой ядерной координате, М - число ядерных степеней свободы, равное ( Зп-6), п - число атомов в молекуле. [15]
Страницы: 1 2 3 4