Вещество - переносенок
Cтраница 2
Для определения окислов никеля, нерастворимых в азотной кислоте, фильтр с веществом переносят в фарфоровый тигель и озоляют в муфельной печи, постепенно повышая температуру до 600 С. После охлаждения остаток растворяют в 5 мл азотной кислоты 1: 1 при кипячении в тигле. Затем раствор выпаривают на кипящей водяной бане досуха. Остаток растворяют в 10 мл 10 % раствора азотной кислоты и далее анализируют, как указано выше. [16]
Лодочку из алюминиевой фольги массой около 1 мг ( см. рис. 4, б) с навеской вещества переносят из камеры для взвешивания ( см. рис. 3, А, слева) в камеру для подготовки образца ( см. рис. 3, А, в центре), где находится аппаратура для приготовления раствора и отбора аликвотной части. [17]
Раствор выпаривается досуха, остаток ( амид тримеллитовой кислоты и следы амида ортофталевой кислоты) переносится в воронку, промывается 15 - 20 мл холодного ацетона или метиловым спиртом ( при этом удаляются следы амида ортофталевой кислоты), после чего вещество переносят на часовое стекло, высушивают до постоянного веса и определяют количество амида тримеллитовой кислоты. Выход амидов ортофталевой и тримеллитовой кислот по методу В. М. Родионова, но нашим экспериментальным данным, полученным с чистыми кислотами, колеблется в пределах 80 - 85 % от теории. Поэтому для выяснения истинного количества амидов тримеллитовой и ортофталевой кислот надо полученное во время опыта количество амидов умножить на 1 25, после чего сделать перс-счет на количество гримеллитовой и ортофталевой кислот. [18]
Смешивают 30 г иодата калия KJO3 и 25 г перекиси натрия Na2O2 в большом фарфоровом тигле и нагревают при темнокрасном калении в течение получаса. Вещество переносят на воронку для отсасывания и вновь промывают водой до тех пор, пока промывная вода не окажется слабо щелочной по фенолфталеину. Препарат высушивают и снова измельчают. Хранят в склянке с притертой пробкой. [19]
Четырехгорлую колбу на 250 мл с мешалкой, обратным холодильником, термометром и капельной воронкой помещают в водяную баню с электрообогревом Загружают 50 мл хлорбензола, нагревают до 30 - 40 С, при перемешивании по каплям добавляют нитрующую смесь, медленно нагревают до 75 С и выдерживают i ч, затем охлаждают до комнатной температуры и постепенно при перемешивании палочкой выливают в стакан на 500 мл, содержащий 200 мл воды. Маслообразное вещество переносят в фарфоровую чашку на 100 мл и сушат в вакуум-эксикаторе над СаСЬ - Безводный нитропродукт охлаждают до 10 - 12 С, при этом 4-нитрохлорбензол закристал-лизовывается и выпадает в осадок. Жидкие продукты, содержащие 2-изомер, декантируют с осадка, а оставшийся 4-изомер пере-кристаллизовывают из 50 мл метилового спирта; 4-нитрохлорбензол кристаллизуется в виде призм или пластинок. [20]
Окисление протекает в твердой фазе. Затем вещество переносят на фильтр, промывают несколько раз горячей водой и сушат на воздухе. [21]
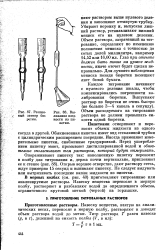 |
Резиновый затвор бюретки.| Вы-ливание жидкости из пи-петкп. [22] |
В мерных колбах ( см. рис. 40) приготовляют титрованные или анализируемые растворы. Навеску вещества переносят в мерную колбу, растворяют и разбавляют водой до определенного объема, ограниченного круговой меткой ( чертой) на горлышке. [23]
 |
Резиновый затвор бюретки.| Выливание жидкости из пипетки. [24] |
В мерных колбах ( см. рис. 39) приготовляют титрованные дли анализируемые растворы. Навеску вещества переносят в мерную колбу, растворяют и разбавляют водой до определенного объема, ограниченного круговой меткой ( чертой) на горлышке. [25]
В подобных случаях часто применяют следующий способ. Навеску вещества переносят в колбу для перегонки и заливают органическим растворителем, не смешивающимся с водой и кипящим без разложения при не очень высокой температуре. Затем: отгоняют часть смеси, причем вместе с ксилолом перегоняется полностью вода из анализируемого вещества. Дистиллат стекает в измерительный цилиндр, в нижней ( более узкой) части которого собирается перегнанная вода. Измерив объем воды, можно рассчитать ее содержание в исследуемом веществе. [26]
В подобных случаях часто применяют следующий способ. Навеску вещества переносят в колбу для перегонки и заливают органическим растворителем, не смешивающимся с водой и кипящим без разложения при не очень высокой температуре. Затем отгоняют часть смеси, причем вместе с ксилолом перегоняется полностью вода из анализируемого вещества. Дистиллат стекает в измерительный цилиндр, в нижней ( более узкой) части которого собирается перегнанная вода. Измерив объем воды, можно рассчитать ее содержание в исследуемом веществе. [27]
Иногда при титровании по остатку, если реакция связана с образованием осадка, применяют следующий прием. Навеску исследуемого вещества переносят в мерную колбу емкостью 100 мл, вливают в нее 50 мл первого рабочего титрованного раствора, затем раствор доводят до метки и перемешивают. Далее фильтруют через сухой фильтр и собирают фильтрат в сухой стакан, причем нет необходимости переносить осадок на фильтр и промывать его. Когда соберется более 50 мл фильтрата, отбирают пипеткой точно 50 мл и титруют вторым рабочим раствором. [28]
Взвешивают около 0 2 - 0 3 г исследуемого эфирата в микробюк-се с пришлифованной пробкой. Бюкс с веществом переносят в коническую колбу, в которой предварительно налито 25 мл раствора ди-бутиламйна в хлорбензоле. [29]
После охлаждения выпадает осадок, который отфильтровывают и сушат. Для удаления бензойной кислоты вещество переносят в стакан, приливают 200 мл абсолютного эфира, тщательно перемешивают стеклянной палочкой и фильтруют. Перекристаллизацию производят из 100 мл воды. [30]
Страницы: 1 2 3