Анализируемое вещество
Cтраница 1
Анализируемое вещество не образует окрашенных перлов. [1]
Анализируемое вещество переводится в As2Og рядом описанных выше последовательных превращений. [2]
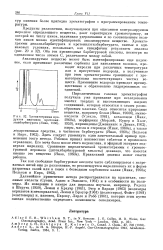 |
Хроматограмма продуктов пиролиза кротонилэтилбарбитурата ( Янак, 1960. [3] |
Анализируемое вещество может быть идентифицировано еще надежнее, если применяют различные сорбенты для наполнения колонок, проводят пиролиз при различных температурах или в присутствии водорода, а полученные при этом хроматограммы сравнивают с хро-матограммами соответствующих чистых веществ. Применение водорода приводит преимущественно к образованию гидрированных соединений. [4]
Анализируемое вещество в сильносолянокислом растворе нагревают в присутствии МпС12 ( катализатор) с избыточным количеством раствора сернокислого гидразина. NaCIO в присутствии метилового красного или индигокармина. [5]
Анализируемое вещество титруют несколько раз с использованием различных индикаторов. Для сравнительной оценки возможностей индикаторов при обнаружении истинной точки эквивалентности следует провести статистическую обработку результатов. Пример такой обработки приведен в гл. [6]
Анализируемое вещество на входе в прибор должно иметь следующие параметры: температура анализируемого вещества на входе в прибор - газов и паров - - 20 - 200 С; жидкостей - 20 - 50 С. [7]
Анализируемое вещество должно быть чистым. Индивидуальность собранной фракции проверяют с помощью ГЖХ. Твердые вещества после проб на растворимость ( см. разд. [8]
 |
Схема хроматографической установки для определения отношения углерод / азот в хроматографически разделенных фракциях. [9] |
Анализируемые вещества разделяют сначала в потоке газа-носителя на хроматографической колонке. Затем они поступают в реактор, где, сгорая, образуют диоксид углерода, воду и азот. Эта смесь сначала проходит через поглотитель, который задерживает воду, и затем через первый детектор, который регистрирует сумму диоксида углерода и азота. После первого детектора поток гелия поступает во второй поглотитель, в котором удаляется диоксид углерода, а затем - во второй детектор, регистрирующий азот. Сравнивая хроматограммы, полученные на обоих детекторах, возможно определить C / N-отношение. [10]
Анализируемое вещество, содержащее от 3 до 900 мкг брома, растворяют в 7 мл воды в конической колбе емкостью 100 мл, снабженной пробкой с двумя отверстиями. Наклоняют колбу под углом 60 для обеспечения максимальной аэрации. В пробку вставлены две трубки; одна из них слегка оттянута и достигает дна наклоненной колбы, другая согнута под углом 60 и присоединена к небольшому барбо-теру, в котором находится поглотитель элементарного брома. По стенке очень медленно добавляют в колбу 2 5 мл концентрированной серной кислоты в течение не менее 10 мин и при постоянном охлаждении водопроводной водой. Вставляют пробку и пропускают воздух 1 час со скоростью около 150 мл / мин. Меняют поглотитель и продолжают аэрацию еще 2 часа. Выделение галоида в течение второго периода аэрации указывает на присутствие слишком больших количеств хлорида, для того чтобы можно было достичь полного разделения этим методом. [11]
Анализируемое вещество должно быть по возможности тонко измельчено. Помещают 5 - 10 г высушенной пробы в платиновую или никелевую чашку и покрывают суспензией Са ( ОН) 2, взятой в количестве, достаточном для поддержания щелочной среды при высушивании и озолении. Высушивание и обугливание проводят на горячей плитке или инфракрасной лампе. Если в золе содержится большое количество кремневой кислоты или если содержание кремневой кислоты неизвестно, то добавляют 5 г гидроокиси натрия и сплавляют смесь, нагревая небольшим пламенем горелки. Плав охлаждают, растворяют в минимальном объеме воды и раствор переносят в дистилляционную колбу при общем объеме воды не более 50 мл. [12]
Анализируемое вещество обрабатывают соляной кислотой, и выделяющиеся газы пропускают через раствор п-амино - М М - диметиланилина в соляной кислоте в колориметрической кювете, затем добавляют раствор FeCl3 и измеряют оптическую плотность. Калибровочную кривую строят по стандартным образцам стали. [13]
Анализируемое вещество, содержащее от 20 до 100 мкг селена, помещают в круглодонную колбу емкостью 100 мл. Добавляют 50 мл этилового спирта для растворения пробы и около 300 мг скелетного никелевого катализатора, свежеприготовленного по методу Дрейка. Присоединяют к колбе вертикальный холодильник и осторожно кипятят 30 мин. После охлаждения сливают спирт с никелевого катализатора и промывают его декантацией 30 мл этилового спирта. Присоединяют к колбе воздушный холодильник и осторожно, по каплям добавляют 4 мл азотной кислоты уд. Колбу нагревают до полного растворения никелевого катализатора. После охлаждения добавляют 10 мл дистиллированной воды и кипятят раствор для удаления окислов азота. [14]
Анализируемые вещества разделяются по колонке с ионообменной смолой, затем проходят через реакционную спираль ( реактор), а флуоресценция регистрируется с помощью флуориметра. Схема модифицированного анализатора аминокислот приведена на рис. 32.11. Для работы на одной колонке необходимо три микронасоса: первый - для подачи буферного раствора на колонку; второй - чтобы довести величину рН элюата до значения 9 и третий предназначен для смешивания элюата с реагентом, представляющим собой раствор флуорескамина в ацетоне. Скорость реакции достаточно высока, что позволяет использовать короткую реакционную спираль. Далее смесь проходит через проточную кювету флуориметра, и интенсивность флуоресценции регистрируется самописцем. Одновременно много внимания было уделено выделению продуктов реакции аминов с флуорескамином, обладающих низкой полярностью, для анализа которых могла оказаться пригодной высокоскоростная хроматография. [15]
Страницы: 1 2 3 4