Скорость - диссоциация
Cтраница 2
Скорость диссоциации доломита меньше, чем магнезита; она увеличивается в присутствии 1 % фторида или хлорида натрия ( рис. 76) 28, 89, эо Обжиг доломита ведут при 700 - 800 или 1100 - 1250 в зависимости от назначения получаемого продукта. При неполном обжиге разлагается лишь MgCO3 и получается так называемый полуобожженный доломит. Его можно, в частности, получить при быстром нагревании доломита: в закрытой зоне вращающейся печи до 750 - 800 в атмосфере СОа в течение 15 мин. [16]
Скорость диссоциации координированных лигандов может изучаться либо методом изотопного обмена, либо спектр офотометриче-ским путем. [17]
Скорость диссоциации координированных лигандов может изучаться либо методом изотопного обмена, либо спектрофотометриче-ским путем. [18]
Скорость диссоциации соответствующих комплексов Со ( П) и Ni ( II) уменьшается точно в обратной последовательности лигандов. [19]
Если скорость диссоциации сопоставима по своей величине со скоростью рацемизации, то возможно, что определяющей скорость стадией как процесса обмена металла, так и рацемизации является гетеролиз одной из связей металла с лигандами; если это не так, то первая стадия состоит в образовании моносольвата, а последующие стадии ( разрыв и синтез) протекают более быстро. В случае правильности этой гипотезы необходимо постулировать, что в некоторых комплексах может происходить изменение типа связи в процессе замещения на воду. Так, в то время как ион [ Fe ( CN) 6 ] 3 - диамагнитен и в нем используются связи 3d24s4p3, гекса-акво-ион [ Fe ( H. Резонанс между ними невозможен; поэтому при замещении на воду должен наступить момент прерывного изменения типа связи. [20]
Поскольку скорость диссоциации этих трех окислов не зависит от давления, равновесие устанавливается очень быстро даже при низких парциальных давлениях в атмосфере. Некоторые окислы азота, например NO3 и N2O6, не могут существовать в атмосфере при нормальных условиях. NO, вероятно, может присутствовать в верхней атмосфере. Только наличие N2O и NO2 было подтверждено измерениями. [21]
 |
Зависимость логарифма константы скорости реакции М2 - 4. [22] |
Однако скорость диссоциации, катализируемая кислотой, остается высокой. Интермедиат с ионом водорода, который замещает ион меди, образуется без дополнительного напряжения каркаса молекулы. [23]
Однако скорость диссоциации метана при этих температурах еще недостаточна и практически для достижения приемлемых выходов водорода процесс приходится вести при температурах 1350 - 1400 С. [24]
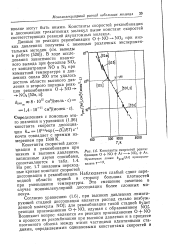 |
Константы скоростей рекомбинации О N0 Аг - NO2 Аг. Пунктирная линия. пекД пропорциональна Т 1 - 9. [25] |
Константы скоростей диссоциации и рекомбинации при низких и высоких давлениях, записанные двумя способами, сопоставляются в табл. 1.4. На рис. 1.7 показаны переходные кривые констант скоростей диссоциации и рекомбинации. Наблюдается слабый сдвиг переходной области кривой в сторону больших плотностей при уменьшении температуры. [26]
Константы скорости диссоциации и рекомбинации можно определять и в том случае, если соответствующая слабая кислота не восстанавливается, однако дает предельные токи разряда ионов водорода, определяемые скоростью диссоциации кислоты. Единственный пока случай такого рода обнаружил Кута [56], исследуя полярографическое поведение борной кислоты. [27]
Уменьшение скорости диссоциации из-за действия растворителя приводит к уменьшению скорости цепных реакций, инициированных примесью веществ и растворенных в том же инертном растворителе. Это положение было выдвинуто Франком и Рабиновичем [22] и применено для расчета инициирования цепных реакций в растворах. Однако это положение правильно лишь для таких инертных растворителей, которые не могут вступать в реакцию со свободными радикалами, образующимися при распаде примеси. Если, как это часто бывает, свободные радикалы в состоянии вступить в реакцию с молеку лами растворителя, то свободная валентность передается частице растворителя и, двигаясь путем эстафетной передачи валентности, может быстро выйти из клетки. Благодаря этому, с одной стороны, скорость инициирования цепей окажется достаточно большой, а, с другой стороны, ускорится процесс рекомбинации свободных валентностей. В такой системе условия перемещения свободной валентности в пространстве близки к поведению свободных радикалов в газовой фазе, с той только разницей, что в газовой фазе радикалы свободно перемещаются в ] пространстве, в то время как в подобного рода растворителях перемещение свободной валентности происходит эстафетным путем от одной молекулы растворителя к другой. [28]
Уменьшение скорости диссоциации из-за действия растворителя приводит к уменьшению скорости цепных реакций, инициированных примесью веществ и растворенных в том же инертном растворителе. Это положение было выдвинуто Франком и Рабиновичем [32] и применено для расчета инициирования цепных реакций в растворах. Однако это положение правильно лишь для таких инертных растворителей, которые не могут вступать в реакцию со свободными радикалами, образующимися при распаде примеси. Если, как это часто бывает, свободные радикалы в состоянии вступить в реакцию с молекулами растворителя, то свободная валентность передается частице растворителя и, двигаясь путем эстафетной передачи валентности, может быстро выйти из клетки. Вследствие этого, с одной стороны, - скорость инициирования цепей окажется достаточно большой, а, с другой, - ускорится процесс рекомбинации свободных валентностей. [29]
Константы скорости диссоциации и рекомбинации можно определять и в том случае, если соответствующая слабая кислота не восстанавливается, однако дает предельные токи разряда ионов водорода, определяемые скоростью диссоциации кислоты. Единственный пока случай такого рода обнаружил Кута [56], исследуя полярографическое поведение борной кислоты. [30]
Страницы: 1 2 3 4