Хинолидное соединение
Cтраница 1
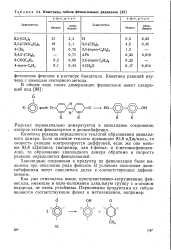 |
Константы гибели феноксильных радикалов. [1] |
Хинолидные соединения и продукты их фенолизации были выделены при окислении ряда фенолов. В условиях окисления диок-сибифенилы могут окисляться далее в соответствующие дифено-хиноны. [2]
Циклогексадиеноны ( хинолидные соединения) в различных реакциях проявляют отчетливо выраженное стремление к превращению в оксиароматические соединения, обладающие более стабильной системой связей. Примером такой реакции является широко известная диенон-фенольная перегруппировка, в процессе которой происходит перемещение одного из геминальных заместителей циклогексадиенона в соседнее положение кольца с образованием замещенного фенола. В соответствии с принятым в настоящее время механизмом, первоначально происходит присоединение протона кислоты к карбонильному кислороду циклогексадиенона с образованием положительно заряженного комплекса I, по-видимому, близкого по структуре а-комплексу. [3]
Хинонитролами называют хинолидные соединения, одним из геминальных заместителей которых является нитро-группа. Эти соединения по своим химическим свойствам очень близки к хиногалбидным соединениям, что, по-видимому, также обусловлено легкостью гемолитического разрыва связи С - NC2 в молекуле хинонитролов. Подобные радикалы могут также возникать при электрохимическом восстановлении хинонитролов 81 или при их пиролизе59 82, приводя к различным продуктам реакции. [4]
Продолжительность существования подобных хинолидных соединений существенно зависит от степени пространственной затрудненности карбонильной группы и при 20 С составляет: в случае R R C ( CH3) 3 - 1.0 суток, R R CH ( CH3) 2 - 4 ч, а при R R CH3 бромхинолидное соединение существует только в разбавленных растворах и при попытке выделения мгновенно изо-меризуется в бромфенол. [5]
Отдельные примеры образования хинолидных соединений из пространственно-затрудненных фенолов в условиях реакций элек-трофильного замещения отмечены и в некоторых других случаях, например, при их хлорировании 42 81 - 91 и азосочетаниим. Однако более подробные данные об этих реакциях в литературе отсутствуют. [6]
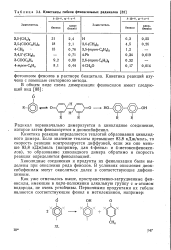 |
Константы гибели феноксильных радикалов. [7] |
Радикал первоначально димеризуется в хинолидное соединение, которое затем фенолизуется в диоксибифенил. [8]
Интересно отметить, что неустойчивые хинолидные соединения получаются при действии азотной кислоты лишь на те 4-замещен-ные 2 6-ди-т / 7ег - бутилфенолы, у которых заместитель в пара-положении обладает сильным отрицательным мезомерным эффектом. При бромировании же различных 2 6-ди-грег - бутилфенолов, содержащих в пара-положении электроотрицательные заместители ( С1, Вг, CN, CHO, COR, COOH, N02), всегда получаются о-хинобромистые соединения 8788 90, которые можно выделить в индивидуальном состоянии. [9]
Наибольшее влияние на процесс образования хинолидных соединений из пространственно-затрудненных фенолов оказывает природа атакующего электрофильного реагента. Переход 2 4 6-три-замещенного фенола в соответствующий циклогексадиенон возможен только под действием сильных электрофильных реагентов. [10]
Как уже отмечалось в предыдущих главах, хинолидные соединения легко образуются при взаимодействии пространственно-затрудненных фенолов с электрофильными и радикальными агентами. Большая часть хинолидных соединений этого ряда, имеющих в качестве геминальных заместителей помимо алкильной группы атомы брома, хлора, гидроксильную, нитро - и другие группы, в индивидуальном виде является устойчивыми и в то же время чрезвычайно реакционноспособными соединениями. [11]
Существенное влияние на направление реакции и устойчивость хинолидных соединений оказывает структура исходного 2 4 6-три-замещенного фенола. Имеются данные, что характер алкильного заместителя мало влияет на скорость процесса образования л-бром-хинолидных соединений при бромировании 2 4 6-триалкилфенолов. Отсутствие в этом случае пространственного влияния грег-бутильной группы на скорость реакции, по-видимому, связано с тем, что при возникновении хинолид-ного соединения нарушается копланарное расположение алкильной группы в связи с изменением гибридизации ( sp2 - - sp3) атома углерода, несущего геминальные заместители. Направление реакции в пара-положение при образовании циклогексадиенонов сохраняется, и при некотором уменьшении электронной плотности ароматического ядра фенола, вызванном введением в орто-положение электроотрицательных заместителей. [12]
Однако в общем случае образующиеся в данных реакциях промежуточные хинолидные соединения должны обладать большей стабильностью, чем соответствующие вещества, получающиеся при замещении атома водорода. Зависимость устойчивости хинолидных соединений от природы геж-заместителей обусловливает отдельные закономерности рассматриваемого процесса, так как прочность связи отщепляющегося заместителя резко влияет на скорость реакции. [13]
При мягком окислении 2 6-ди-грег - бутилфенола окисью серебра в растворе бензола14 получено хинолидное соединение XII, которое является таутомером бис-фенола XIII. Хинолидное производное XII достаточно устойчиво в неполярных растворителях, но в полярной среде быстро превращается в соединение XIII. Аналогично происходит и димеризация феноксильного радикала, получающегося при окислении 2 4-ди-грет - бутилфенола феррицианидом калияш. В этом случае реакция останавливается на стадии образования 2 2 -ди-окси - 3 3 5 5 -тетра-трег - бутилдифенила. [14]
Появляющиеся в ходе ингибированного окисления феноксиль-ные радикалы могут реагировать с перекисными радикалами, давая соответствующие перекидные хинолидные соединения. [15]
Страницы: 1 2 3