Трифтор
Cтраница 2
Типичным примером является фторирование 1 2 4-трихлор-бензола смесью комплексов фтористого нитрозила и фтористого водорода. Образуются как трифтор -, так и дифторпроизводное. По-видимому, кроме того, получающийся в ходе реакции хлористый нитрозил вызывает дополнительно хлорирование, так как образуются также дихлордифтор - и трихлорфторбензолы. Около 10 % вступившего в реакцию трихлорбензола подвергается пер-галогенированию и расщепляется, давая четырехфтористый углерод, хлортрифторметан и дихлордифторметан. [16]
Кварцевую трубку длиной 50 - 70 см и диаметром 0 8 - 1 0 см нагревают в трубчатой печи до 650 - 700 С. Димер бис ( трифтор метил) тиокетена медленно в токе инертного газа подают в зону нагрева. Бис ( трифторметил) тиоке-тен собирают в два последовательно соединенных приемника, охлаждаемых до 0 С. Бис ( трифторметил) тиокетен может длительное время храниться в стеклянной таре, не претерпевая заметной ди-меризации, однако даже следы реагентов основного характера вызывают быструю димеризацию. [17]
 |
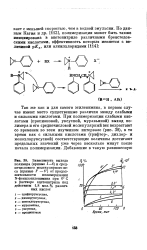
Усредненные характеристики связей [ 1, р. 21 ].| Энергия диссоциации связей. [18] |
Из табл. 3.7 следует, что за исключением фтора для всех галогенов важную роль играют стерические факторы, а из табл. 3.8 видно, что существует четкая зависимость в энергии диссоциации при переходе от фтора к иоду. Сравнение энергии диссоциации связей в трифтор - и трихлорметильных производных также указывает на роль стерических факторов. В самом деле, между характеристиками связей метильной и трифторметильной группы существует большее соответствие, чем между характеристиками связей метильной и трихлорметильной группы; в последнем случае стерические факторы, по-видимому, играют большую роль. Действительно, именно пространственные затруднения ограничивают устойчивость перхлоралканов, где силы отталкивания быстро снижают энергию диссоциации связей С-С по мере роста длины углеродной цепи. По этой причине не удалось получить полиперхлорэтилен ( ССЬ-CClz) n в то время как полиперфторэтилен ( CF2 - CF2) n существует и очень устойчив. [19]
В облученном политетрафторэтилене наблюдались процессы термолюминесценции и фосфоресценции, но они не очень ярко выражены. Сходство полученных спектральных данных с таковыми для трифтор ацетон а свидетельствует о том, что весь наблюдаемый эффект может быть обусловлен карбонильными группами, присутствующими в качестве примесей. С, которые отчасти согласуются, например, с данными по сильному возрастанию молекулярной подвижности, в частности с появлением максимумов упруговязких потерь. [20]
При фторировании фторангидридов кислот гможет быть достигнут высокий выход. Так, например, из ацетилфторида может образоваться трифтор ацетил фторид с 85 % - ным выходом. Хотя с увеличением углеродной цепи выходы несколько падают, они всегда приблизительно в 3 раза больше выходов, получаемых при непосредственном фторировании собственно карбоно-вых кислот. При фторировании фторангидрида н-масляной кислоты, наряду с 36 % фторангидрида н-перфтормасляной кислоты, образуется также около 4 % фторангидрида перфторпро-пионовой кислоты. При электрохимическом фторировании га-логенангидридов карбоновых кислот, как и самих кислот, всегда происходит подобная деструкция. В самом деле следует ожидать образования целого ряда фторангидридов перфтор-кислот ( от трифторацетилфторида до перфторированного фтор-ангидрида исходной кислоты), причем вследствие радикальных рекомбинаций могут получиться фторангидриды высших и даже многоосновных перфорированных кислот. [21]
Влияние большинства протонных кислот сводится к замедлению процесса поляризации молекул а-цианакрилатов, чем и объясняется их применение в качестве ингибиторов. Исключение составляют лишь наиболее сильные кислоты - соляная, фтористоводородная, фосфорная, трифтор - и циануксусные и, прежде всего, концентрированная серная и ее органические эфиры. [22]
Некоторые метилкетоны были проацилированы этиловыми эфирами алкокси - или галоидозамещенных уксусных кислот с образованием соответствующих алкокси - или галоидозамещенных р-дикетонов. Так, например, ацетон ацилируется этиловым эфиром этоксиуксусной кислоты, а ацетон и трифтор ацетон - этиловым эфиром трифторуксуснрй кислоты с хорошими выходами. [23]
В литературе описаны два других общих примера превращения дисульфидов в сульфиды. Брандт, Эмелеус и Хасселдин58 показали, что под действием ультрафиолетового облучения из бис - ( трифтор ( метил) - дисульфида образуется некоторое количество бис - ( трифторметил) - сульфида, опять-таки по свободнора-дикальному механизму. [24]
Обратный порядок - ацилирование свободных аминокислот трифторуксусным ангидридом в безводной трифторуксусной кислоте [117] - не подходит, особенно для количественного получения этих соединений. При ацилиро-вании свободных аминокислот тиоэтиловым эфиром трифторуксусной кислоты в щелочном растворе получают самые различные выходы [14], причем диаминокислоты Лиз и Орн дают только s - N-ТФА-производные, и, следовательно, их можно трифтор ацетил и-ровать по а-положению с помощью других методов. [25]
Так же как и для самого этиленимина, в первом случае имеют место существенные различия между слабыми и сильными кислотами. При полимеризации слабыми кислотами ( пропионовой, уксусной, муравьиной) выход полимера и его среднечисловои молекулярный вес возрастают со временем во всем изученном интервале ( рис. 38), в то время как с сильными кислотами ( трифтор -, дихлор - и монохлоруксусной) соответствующие величины достигают предельных значений уже через несколько минут после начала полимеризации. [27]
В реакциях соединений элементов группы VA часто требуется сделать выбор между двумя альтернативными механизмами, из которых один включает промежуточное образование арсониевой соли, а второй представляет собой гемолитическое замещение у атома мышьяка. Хотя обычно выбор делается в пользу ониевого промежуточного соединения, экспериментальные Доказательства единичны. Реакция бис - ( трифтор гаетид) нитроокиси представляет собой весьма специфический случай, однако, поскольку сама нитроокись является свободным радикалом, то образование промежуточного радикала 5 практически не вызывает сомнений. [28]
Ранние исследования позволили установить, что можно определять количества хрома уже порядка 2 10 - 12 г. Предел детектирования определяется наличием примесей в растворителе ( толуоле), а не шумом или неспособностью детектора откликаться на меньшие концентрации комплекса. Сродство хелатов к электрону определяется как ионом металла, так и степенью галогенирования лиганда. Детектор значительно более чувствителен к трифтор - и гексафтор-ацетилацетонатам, чем к аналогичным ацетилацетонатам. То же наблюдается и при детектировании алюминиевых комплексов тех же трех лигандов. Сигнал детектора сильно зависит и от природы иона металла; при заданной концентрации трифторацетил-ацетонат хрома ( Ш) дает приблизительно в 10 раз больший сигнал, чем соответствующий комплекс алюминия. [29]
При фторировании трихлорсилана SnF4 и TiF4 ими были получены трифтор -, хлордифтор - и дихлорфторсиланы. Однако при фторировании трихлорсилана SbF3 и AsF3 единственным продуктом реакции был четырех-фтористый кремний. Позднее [3, 4] было установлено, что при фторировании трихлорсилана фтористой сурьмой в присутствии SbCl6 можно получить моно -, ди - и трифтор замещенные продукты. [30]
Страницы: 1 2 3