Двухэлектронный интеграл
Cтраница 3
Этот результат означает, что кулоновские интегралы не должны зависеть от ориентации р-орбиталей. Поэтому Попл с сотрудниками ввел предположение, что кулоновские двухэлектронные интегралы ( nujvv) зависят только от природы атомов, на которых локализованы орбитали / ц, и % v, и не зависят от конкретного вида орбиталей. [31]
Этот результат означает, что кулоновские интегралы не должны зависеть от ориентации р-орбиталей. Поэтому Попл с сотрудниками ввел предположение, что кулоновские двухэлектронные интегралы ( mi vv) зависят только от природы атомов, на которых локализованы орбитали Хц и XY, и не зависят от конкретного вида орбиталей. [32]
Проблема недаагональных множителей не возникает. В этом случае выражение для среднего значения энергии включает помимо кулоновских и обменных интегралов более сложные двухэлектронные интегралы, но зависимость коэффициентов перед ними от орбиталей становится аналитической. [33]
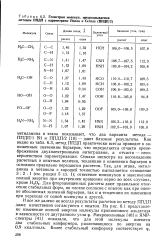 |
Геометрия молекул, предсказываемая методом ППДП с параметрами Попла и Сегала ( ППДП / 2. [34] |
ППДП / 1 [9] и ППДП / 2 [10] - дают близкие результаты. Как видно из табл. 6.3, метод ППДП практически всегда приводит к заниженным значениям барьеров, что определяется отчасти пренебрежением двухэлектронными интегралами, а отчасти - несовершенством параметризации. Однако несмотря на несовпадение расчетных и опытных величин, тенденции в изменении барьеров в основном правильно передаются расчетом. Так, для этана, метиламина и метанола с хорошей точностью выполняется отношение 3: 2: 1 ( см. раздел 5 гл. Для фторзамещенных расчеты дают правильное отнесение стабильных форм, но плохое согласие с опытом как при оценке абсолютных значений барьеров, так и в тенденции их изменения при переходе от одной молекулы к другой. [35]
Как видим, исследование многоэлектронной проблемы с проведением непосредственных вычислений, необходимых матричных элементов, очевидно, чрезвычайно трудоемко. Эти вычисления можно, конечно, как-то систематизировать, если использовать некоторые общие правила, позволяющие непосредственно сводить рассматриваемые матричные элементы к одно - и двухэлектронным интегралам. В методе Слейтера это как раз и делается, в нем волновая функция представляется как линейная комбинация детерминантов и выводится общее выражение для типичного матричного элемента между типичной парой детерминантов. [36]
Необходимо подчеркнуть, что, кроме диаграмм, приведенных на рис. 3 и 4, существует ряд других диаграмм, дающих вклад в оба последовательных механизма. Числа, приведенные на рис, 5 рядом с каждой диаграммой, характеризуют относительный вклад данной диаграммы по сравнению с диаграммой d на рис. 3 в предположении, что все двухэлектронные интегралы равны и что энергии перехода, соответствующие одинарному замещению орбиталей, равны 1 / 2 энергии перехода, соответствующей двойному замещению, и 1 / 3 энергии тройного замещения. [37]
Однако в методе ВКВЛО - ППДП / 2 условие ортогональности в явном виде не вводится, и поэтому истинная природа базисных орбиталей не совсем ясна. Они могут рассматриваться как усеченные орбитали при расчете перекрестных плотностей ( cpi, TNPJ), но не при расчете матричных элементов оператора Фока ( фг, Ftyi) - Поэтому расчет двухэлектронных интегралов в соответствии со схемой ППДП / 2 требует, кроме введения гипотезы усеченных орбиталей, ряда дополнительных предположений. [38]
Кроме того, привлечение свойств молекулярной симметрии позволяет построить фоковскую матрицу в блок-диагональной форме, для которой проблема собственных значений может быть решена последовательно блок за блоком. При проведении молекулярных расчетов объем вычислений на стадии определения интегралов зависит от четвертой степени числа базисных функций, на стадии построения фоковской матрицы - также от четвертой степени числа базисных функций, а на стадии диагона-лизации этой матрицы - от третьей степени числа базисных функций. Стадия преобразования двухэлектронных интегралов к базисному набору двухэлектронных интегралов по молекулярным орби-талям зависит от пятой степени числа базисных функций, но и в этом случае учет молекулярной симметрии позволяет существенно снизить объем вычислений, необходимых для осуществления этого преобразования. Естественно, наибольшая эффективность достигается при условии, что базисные функции и молекулярные орбитали симметризованы относительно точечной группы симметрии молекулы. [39]
Недостатки (2.3) по сравнению с (2.1) известны: неправильная асимптотика вблизи ядра атома и при г - оо. Вследствие этого более или менее корректное описание хартри-фоковских АО с помощью линейных комбинаций гауссовских функций требует в 2 - 5 раз большего числа слагаемых, чем при использовании разложений по слейтеровским функциям. Это приводит к значительному увеличению количества двухэлектронных интегралов, вычисляемых с гауссовскими функциями. [40]
Кроме того, привлечение свойств молекулярной симметрии позволяет построить фоковскую матрицу в блок-диагональной форме, для которой проблема собственных значений может быть решена последовательно блок за блоком. При проведении молекулярных расчетов объем вычислений на стадии определения интегралов зависит от четвертой степени числа базисных функций, на стадии построения фоковской матрицы - также от четвертой степени числа базисных функций, а на стадии диагона-лизации этой матрицы - от третьей степени числа базисных функций. Стадия преобразования двухэлектронных интегралов к базисному набору двухэлектронных интегралов по молекулярным орби-талям зависит от пятой степени числа базисных функций, но и в этом случае учет молекулярной симметрии позволяет существенно снизить объем вычислений, необходимых для осуществления этого преобразования. Естественно, наибольшая эффективность достигается при условии, что базисные функции и молекулярные орбитали симметризованы относительно точечной группы симметрии молекулы. [41]
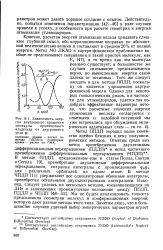 |
Зависимость энергии внутреннего вращения молекулы пропионового альдегида от двугранного угла ф. [42] |
Того же следует ожидать и для метода ППДП, поскольку он является упрощением хартри-фоковской модели. Метод ППДП породил целое семейство идейно близких к нему полуэмпирических методов. В методе ПДДП, предложенном еще в статье Попла, Сантри и Сегала [9], пренебрегают двухатомным дифференциальным перекрыванием, сохраняя интегралы с произведениями q cpv для различных орбиталей на одном атоме. В методе ЧПДП [11] удерживают все одноцентровые обменные интегралы, пренебрегая остальными двухэлектронными интегралами. Мы не будем анализировать точность предсказания геометрии молекул и конформационных энергий, которую дают разные методы этого типа. [43]
Любой интеграл вида / 5 i) / отличается от нуля только в том случае, если в произведении неприводимых представлений Гг X Го X Г / содержится полносимметричное неприводимое представление. Оператор Гамильтона, а также его различные части являются полносимметричными. Орбитали симметричны относительно отражения в молекулярной плоскости, а я-орбитали антисимметричны. Следовательно, произведение ГаХГя антисимметрично по крайней мере относительно этой операции и поэтому не может быть полносимметричным. Однако двухэлектронные интегралы могут включать две 0-функции и две я-функции. Эти интегралы могут отличаться от нуля, если две а-функции и две я-функции совпадают либо если каждая из а - и я-функций имеет одинаковое поведение относительно всех генераторов, за исключением молекулярной плоскости симметрии. [44]
Страницы: 1 2 3