Вычислительное время
Cтраница 2
Для задачи назначения большой размерности, а также при лимитированном расходе вычислительного времени часто приходится прибегать к приближенным методам. Приведем некоторые из них, наиболее распространенные. [16]
Если, однако, из-за большого вычислительного времени ( алгоритмы координирования требуют много вычислительного времени) или из-за малости ожидаемого экономического эффекта применение специальных алгоритмов кородинирования невозможно, то рекомендуется измерять величины д; /, и y f, /, и подавать в программу управления. Такой подход рекомендуется и тогда, когда математическая модель одной подсистемы, которая включается в координирование, недостаточно адекватна реальному химико-технологическому процессу, чтобы осуществить оптимизацию только с помощью математической модели. [17]
Эти полуэмпирические методы, как их принято называть, намного проще неэмпирическнх и требуют гораздо меньших затрат вычислительного времени. Поэтому они применимы к еще большим молекулам. Их важнейшим недостатком является непредсказуемая точность. [18]
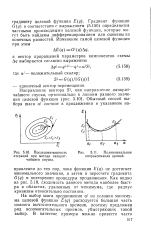 |
Последовательность итераций при методе наикратчайшего спуска. [19] |
На выбор шага продвижения а3 из условия минимума целевой функции E ( q) расходуется большая часть полного вычислительного времени, поэтому предложен ряд вспомогательных приемов, убыстряющих такой выбор. [20]
Принимая во внимание, что в задачах такого типа k гораздо меньше п, видим, что экономия вычислительного времени зна. [21]
Недостаток метода - необходимость довольно точного определения минимума целевой функции на каждой итерации, что приводит к увеличению затрат вычислительного времени. С другой стороны, здесь не требуется обращение матрицы. Реализация алгоритма не предъявляет больших требований к памяти ЭВМ. [22]
Другими словами, преимущество алгоритма Гаусса при редукции системы уравнений с симметричной матрицей коэффициентов заключается в том, что можно сэкономить почти половину вычислительного времени, необходимого для редуцирования системы уравнений с несимметричной матрицей. [23]
В 1960 - е годы введение базисов гауссовых функций для молекулярных расчетов ( основанное на предложении С. Ф. Бойза, сделанном в 1950 г.) значительно снизило вычислительное время, необходимое для получения хороших результатов при хартри-фоковских расчетах молекул, что сделало реальными расчеты больших молекул. Развиты и продолжают развиваться различные методы хотя бы частичной компенсации корреляционной ошибки. Полный расчет по методу конфигурационного взаимодействия с применением функций, определяемых выбранным базисным набором, в принципе должен исключить всю корреляционную ошибку, которую можно учесть при использовании данного базисного набора; однако проблема быстро становится практически неразрешимой при возрастании размеров системы. По этой причине расчеты по методу конфигурационного взаимодействия ( KB) проводятся лишь с учетом ограниченного числа конфигураций. В последнее время разработаны многоконфигурационные методы ССП, в которых волновые функции возбужденных конфигураций оптимизируются одновременно с оптимизацией функции основного состояния. Эти и многие другие усовершенствования призваны постоянно повышать точность молекулярных расчетов. Тем временем удается непрерывно получать полезные результаты с использованием уже отработанных методов. [24]
При использовании в АСУП ЭВМ третьего поколения часто применяют схемы обработки, связанные с предварительной записью на магнитные ленты или диски информации, подготовленной на перфокартах или перфолентах, что позволяет экономить дорогостоящее вычислительное время машины. [25]
При использовании в ИВЦ АСУ ЭВМ третьего поколения часто применяют схемы обработки, связанные с предварительной записью на магнитные ленты или диски информации, подготовленной на перфокартах или перфолентах, что позволяет экономить дорогостоящее вычислительное время машины. [26]
Для метода Якоби параметр 0 определяется экспериментально. Вычислительное время пропорционально числу итераций метода Якоби, требуемых для получения желаемой точности. [27]
Они требуют больших затрат вычислительного времени и памяти, но сходятся за меньшее число итераций. [28]
Это разделение производится с целью определения таких классов, чтобы для каждого из них применялись свои структуры или свои параметры алгоритма управления. Тем самым удается сильно сократить вычислительное время, необходимое для определения оптимальных управляющих воздействий. [29]
Это делает процедуру намного проще, чем решение уравнений (4.20) и (4.21) относительно п 1 переменной. Кроме того, чтобы сэкономить вычислительное время, предлагается вначале процедуры одновременно привести к диагональному виду ковариационные матрицы Si и 1г и далее работать в преобразованной системе координат. [30]
Страницы: 1 2 3 4