Интерпретация - электронный спектр
Cтраница 2
Речь идет о конфигурационном взаимодействии первого порядка между вырожденными функциями, без учета которого интерпретация электронного спектра практически неосуществима. В рамках метода ограниченного конфигурационного взаимодействия ( ОКБ) Паризера - Парра предполагается наличие взаимодействия между определенным числом однократно возбужденных конфигураций. Если для вычисления, матричных элементов KB использовать коэффициенты разложения МО ССП, можно показать ( см. разд. [16]
Речь идет о конфигурационном взаимодействии первого порядка между вырожденными функциями, без учета которого интерпретация электронного спектра практически неосуществима. В рамках метода ограниченного конфигурационного взаимодействия ( ОКБ) Паризера - Парра предполагается наличие взаимодействия между определенным числом однократно возбужденных конфигураций. Если для вычисления матричных элементов KB использовать коэффициенты разложения МО ССП, можно показать ( см. разд. [17]
Кроме различных видов химических формул, в число качественных моделей входят электронные формулы, возникшие при интерпретации электронных спектров. В спектроскопии разработан особый язык, на котором выражаются эти формулы. [18]
Соединения, характеризующиеся поглощением в ближнем ультрафиолете, обычно дают достаточно интенсивные пики молекулярных ионов, что позволяет вычислять необходимые для интерпретации электронных спектров молярные коэффициенты поглощения. [19]
Из приведенных выше данных расчета большого числа молекул следует, что хкжкелевскпе орбитали с конфигурационным взаимодействием дают, если соответствующие матричные элементы вычислены должным образом, хорошую интерпретацию электронного спектра. Теперь необходимо сравнить величины, определенные этим методом, с величинами, полученными па основе ССП-молекулярных орбпталеи с включением конфигурационного взаимодействия. [20]
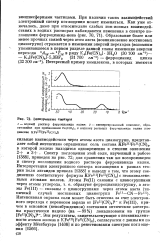 |
Электронные спектры. [21] |
Спектр поглощения этой соли, изученный в работе [1388], приведен на рис. 72; для сравнения там же воспроизведен и спектр поглощения водного раствора ферроцианида калия. Интерпретация электронного спектра поглощения в рамках теории поля лигандов привела [1388] к выводу о том, что этому соединению соответствует формула KFein [ FeII ( CN) 6 ] с неэквивалентными атомами железа. [22]
Рассмотренные в разделе методы электронной спектроскопии в видимой и УФ областях являются основными источниками экспериментальных данных об энергетических состояниях молекул, связанных с электронным возбуждением. Интерпретация электронных спектров молекул основывается на использовании представлений приближенных методов квантовой химии. С другой стороны, получаемые из спектров данные могут служить критерием пригодности и ограничений тех или иных приближений в описании электронных состояний и переходов, стимулирующим развитие и уточнение теоретических представлений. [23]
Найдено, что значения интенсивности поглощения, вычисленные с помощью квантовомеханических расчетов, по порядку величины совпадают с наблюдаемыми значениями. Для интерпретации электронных спектров соединений с сопряженными связями и ароматических соединений успешно применяются квантовомеханическая модель свободного электрона и простой метод МО. Рост цепи сопряжения в циклических и линейных сопряженных системах обычно сопровождается батохромным сдвигом полосы поглощения, однако это нельзя считать общим правилом. Направление сдвига часто удается предсказать на основании вычисленной разности между энергиями высшей занятой и низшей свободной МО. Простая теория МО позволяет удовлетворительно объяснить электронные спектры комплексов с переносом заряда. Для более строгой интерпретации электронных переходов необходимо применять усовершенствованные методы расчета, позволяющие учитывать конфигурационное взаимодействие. [24]
Как показывает анализ спектра третичного бутилбензола, это также противоречит опыту. По-видимому, для интерпретации электронных спектров моноалкилбензолов нужно сразу отказаться от модели молекулы с симметрией Cs2 Действительно, согласно правилам отбора, основные переходы А1 - А1 и Al - - A1 - At в молекуле с С5а, соответствующие в группе симметрии C2v переходам А - В и А1 - В1 - В, неразличимы по поляризации, что также не согласуется с экспериментом. [25]
Этот раздел относительно краток по нескольким причинам, однако они ни в коей мере не связаны с тем, что недавно был опубликован [30] обзор по электронным спектрам адсорбированных молекул. Тот факт, что интерпретация электронных спектров ( адсорбированных молекул) большей частью осуществляется путем сравнения с известными спектрами и с известными спектральными изменениями в различных растворителях, а не на основе более надежных расчетов электронных энергетических уровней, которые весьма сложны, позволяет сделать раздел, посвященный интерпретации электронных спектров, относительно кратким. Кюветы для регистрации электронных спектров можно изготовлять из простого или плавленого кварца, легкодоступного и обладающего прозрачностью в видимой и ближней ультрафиолетовой спектральных областях, по этим причинам конструкция УФ-кюветы оказывается более простой, чем конструкция ИК-кю-веты. [26]
В литературе имеется - несколько различных, систем обозначений полос поглощения в соответствии с детализацией типов электронных переходов, учитывающих симметрию молекулярных орбиталей. Эта усложненная символика предназначается для углубленной интерпретации электронных спектров соединений точно известной структуры. При рутинном структурном анализе она используется редко и в рамках данного руководства мы ограничились простейшей системой обозначений, предложенной Бура-вым и Брауде более 40 лет тому назад. [27]
В нулевом приближении пр - и ns - О рбиты фтора и ксенона и орбиты ксенона типа d могут считаться несвязывающими. Позже мы покажем, что эффекты я-связывания важны при интерпретации электронных спектров и потенциалов ионизации фторидов ксенона. [28]
Расчеты электронных спектров в параметризации CNDO / 2 с учетом однократно возбужденных конфигураций приводят к плохому согласию с наблюдаемыми величинами энергий переходов. Такая ошибка в вычислении энергий перехода делает непригодным метод CNDO / 2 для интерпретации электронных спектров, в то время как геометрическая структура возбужденных состояний ( пример СН2) может описываться вполне удовлетворительно. [29]
Расчеты электронных спектров в параметризации CNDO / 2 с учетом однократновозбужденных конфигураций приводят к плохому согласию с наблюдаемыми величинами энергий переходов. Такая ошибка в вычислении энергий перехода делает непригодным метод CNDO / 2 для интерпретации электронных спектров, в то время как геометрическая структура возбужденных состояний ( пример СН2) может описываться вполне удовлетворительно. Этот результат является прямым следствием параметризации резонансных интегралов 3рт под свойства основных состояний молекул. [30]
Страницы: 1 2 3