Интерпретация - хроматограмма
Cтраница 2
Исследование точности предложенного ранее метода кол-венной интерпретации хроматограмм смесей, содержащих компонент, идентичный газу-носителю. [16]
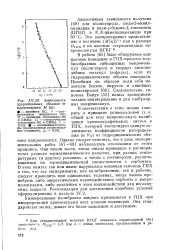 |
Зависимость удерживаемых объемов от произведения М [ ц ]. [17] |
Универсальная калибровка широко применяется в ГПХ при интерпретации хроматограмм всех классов полимеров. Она стандартизует метод, делает его более надежным и доступным. [18]
Янак [554] провел статистическое сравнение различных методов интерпретации хроматограмм, применяя лишь метод нормировки. [19]
Для выяснения точности анализа при различных методах интерпретации хроматограмм одним из авторов совместно с В. Т. Шемя-тенковой, Н. А. Паламарчук и Л. А. Нечаевой [516] был проведен хроматографическнй анализ смеси бензол - гексан. [20]
Однако при ШХ-анализе ВМС Н следует проявлять известную остодожность в интерпретации хроматограмм, полученных с помощью этих детекторов. Так, при ГПХ-разделе-нии полимеров обычно предполагают, что сигнал детектора для различных гомологов зависит только от их концентрации. В случае же анализа тяжелых нефтяных фракций необходимо учитывать, что и показатели преломления и оптическая плотность прежде всего зависят от химической структуры вещества. Игнорирование этого обстоятельства при ГПХ-анализе ВМ СН может привести к неправильной интерпретации полученных данных. Возможными путями решения этой проблемы являются либо применение пламенно-ионизационного ( транспортного) детектора, либо коррекция сигнала детектора путем гравиметрического определения количества вещества в отобранных фракциях. [21]
С каждым из этих трех параметров могут применяться ч етыре метода интерпретации хроматограммы: нормировка, внутренняя стандартизация, нормировка с введением калибровочных коэффициентов и абсолютная калибровка. [22]
Несмотря на то, что точность определения состава при различных методах интерпретации хроматограмм является важным вопросом, в литературе имеется относительно мало работ в этой области. [23]
Таким образом, имеются различные в ряде случаев противоречивые точки зрения на точность интерпретации хроматограмм различными методами. [24]
К недостаткам метода следует отнести большой расход газа-носителя, периодичность анализа и трудность интерпретации хроматограмм. [25]
При изотермической хроматографии температура колонки существенно влияет на разделение смеси, длительность анализа и возможность интерпретации хроматограмм. При выборе оптимальной температуры колонки приходится находить компромиссное решение, обеспечивающее как приемлемую длительность анализа и возможность точного расчета хроматограммы, так и удовлетворительное разделение компонентов. Оказалось, что для данного вещества эта идеальная рабочая температура обычно близка к его температуре кипения. Поэтому температура колонки должна находиться в середине интервала температур кипения компонентов смеси. Если этот интервал охватывает более 100, то средняя температура колонки слишком резко отличается от оптимальной температуры для крайних компонентов и пики этих компонентов трудно, а иногда и вообще невозможно рассчитать. Низкокипящие компоненты очень быстро выходят из колонки и при этом плохо разделяются, тогда как высококипящие, напротив, слишком длительное время удерживаются колонкой и дают на хроматограмме очень растянутые пики. Таким образом, может оказаться, что отдельные компоненты вообще не будут определены. [26]
При изотермической хроматографии температура колонки существенно влияет на разделение смеси, длительность анализа и возможность интерпретации хроматограмм. При выборе оптимальной температуры колонки приходится находить компромиссное решение, обеспечивающее-как приемлемую длительность анализа и возможность точного расчета хроматограммы, так и удовлетворительное разделение компонентов. Оказалось, что для данного вещества эта идеальная рабочая температура обычно близка к его температуре кипения. Поэтому температура колонки должна находиться в середине интервала температур кипения компонентов смеси. Если этот интервал охватывает более 100, то средняя температура колонки слишком резко отличается от оптимальной температуры для крайних компонентов и пики этих компонентов трудно, а иногда и вообще невозможно рассчитать. Низкокипящие компоненты очень быстро выходят из колонки и при этом плохо разделяются, тогда как высококипящие, напротив, слишком длительное время удерживаются колонкой и дают на хроматограмме очень растянутые пики. Таким образом, может оказаться, что отдельные компоненты вообще не будут определены. [27]
Применение высокоэффективной газовой хроматографии при анализе сложных смесей не всегда возможно из-за отсутствия сравнительных данных, необходимых для интерпретации хроматограмм. В настоящей работе показано, что проведение реакции метиленирования в сочетании с капиллярной газовой хроматографией является новым удобным методом синтеза и измерения времен удерживания многих углеводородов. [28]
Применение высокоэффективной газовой хроматографии при анализе сложных смесей не всегда возможно из-за отсутствия сравнительных данных, необходимых для интерпретации хроматограмм. В настоящей работе показано, что проведение реакции метиленировэния в сочетании с капиллярной газовой хроматографией является новым удобным методом синтеза и измерения времен удерживания многих углеводородов. [29]
Для каждого из трех параметров, характеризующих содержание компонента в смеси, может быть применен один из следующих четырех методов интерпретации хроматограммы: внутренняя нормализация, внутренняя стандартизация, нормализация с введением калибровочных коэффициентов, абсолютная калибровка. [30]
Страницы: 1 2 3 4