Вычисление - термодинамическая функция
Cтраница 3
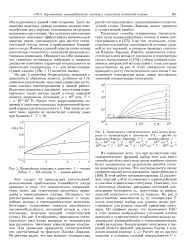
Из сказанного ясно, что при вычислении термодинамических функций выбор того или иного способа расчета статсуммы атома наиболее сильно будет сказываться на результатах расчета степени ионизации плазмы. В этой работе проанализированы 12 различных моделей учета кулоновского взаимодействия и способов ограничения статсуммы. Показано, что в некоторых областях диаграммы состояний концентрация электронов пе, полученная из разных моделей отличается почти на порядок. На рис. 4 представлены зависимости пе от температуры Т вдоль некоторых изобар для плазмы цезия, рассчитанные для разных моделей уравнения состояния. Из графика видно, что применение ПБС и учет высоко возбужденных состояний атомов приводит к существенному ослаблению вклада кулоновского взаимодействия в термодинамические свойства плазмы. [32]
Из сказанного следует, что при вычислении термодинамических функций растворения с использованием хроматографических данных следует вводить поправку на отклонение от идеальности паровой фазы данного вещества, особенно при высоких давлениях насыщенного пара. [33]
В главе II настоящего Справочника рассмотрены методы вычисления термодинамических функций идеальных газов по молекулярным постоянным. Рассчитанные таким образом термодинамические функции газов достаточно точны в области высоких температур и низких давлений. При низких температурах и высоких давлениях термодинамические функции газов, вычисленные без учета межмолекулярного взаимодействия, могут значительно отличаться от термодинамических функций соответствующих реальных газов. В настоящем Приложении рассматривается методика учета межмолекулярных взаимодействий при вычислении термодинамических функций газов; приводятся р - V - Г - данные и значения силовых постоянных межмолекулярного потенциала некоторых, рассматриваемых в Справочнике газов, для которых известны экспериментальные р - V - Т - данные. [34]
Ниже мы рассматриваем в общих чертах метод вычисления термодинамических функций совершенных газов на основании суммы состояний, данный Жио-ком 2 ] в 1930 г. Рассматриваемые уровни энергий и суммы состояний относятся к соответствующим внутренним степеням свободы. [35]
Следует отметить, ЧТОБ работах [2940, 2357, 4345, 1516, 420] при вычислении термодинамических функций SO применялись разные методы и использовались разные значения физических постоянных. Наиболее близкими к настоящему Справочнику по методу вычислений и принятым значениям физических постоянных являются термодинамические функции SO, приведенные в первом издании Справочника. Различие в значениях Фг в первом и в настоящем изданиях Справочника составляет 0 032, 0 003 и 0 258 кал / моль-град при Т - 298 15, 3000 и 6000 К, соответственно. При низких температурах это различие обусловлено преимущественно различием в принятых значениях молекулярных постоянных, при высоких температурах - учетом в настоящем издании Справочника ряда возбужденных электронных состояний SO с относительно небольшими энергиями возбуждения. [36]
Причины успеха одновременного использования обоих приближений при вычислении термодинамических функций дебаевской плазмы следующие. Во-вторых, недостаток приближения парных столкновений в том, что оно плохо воспроизводит вклад взаимодействия на больших расстояниях, но его вклад на этих расстояниях мал, а поляризационное приближение наоборот - плохо воспроизводит вклад взаимодействия на малых расстояниях, где относительно мал его вклад. Вклад этих ошибок растет с плотностью зарядов. [37]
За последнее время был достигнут значительный прогресс в вычислении термодинамических функций непосредственно из суммы состояний для некоторых веществ, по поведению приближающихся к идеальному газу. Однако вычисление термодинамических функций для реальных газов и жидкостей затруднено из-за отсутствия сведений о межмолекулярных силах. Изменение термодинамических функций реальных газов и жидкостей наиболее удобно вычислять с помощью эмпирических уравнений для макроскопических свойств или эмпирического уравнения состояния. Для количественного вычисления необходимо выразить термодинамические функции в зависимости от измеримых макроскопических свойств, таких как давление, объем, температура, теплоемкость и состав. [38]
Возбужденные электронные состояния молекул HD и Da при вычислении термодинамических функций не учитывались, так как их вклад при Т 6000 К весьма мал. [39]
В настоящем Справочнике и в работе [1273] при вычислении термодинамических функций ССЬВг2 использованы одинаковые значения основных частот. Приведенные в работе [1273] значения Ф т и S T ниже соответствующих значений этих величин в табл. 184 ( II) примерно на 0 04 кал / моль - град. Имеющиеся расхождения обусловлены в основном различием принятых значений главных моментов инерции. В работе [1273] они были вычислены для Гс-с - 1 75 и ГС-БГ 1 93 А, тогда как в настоящем Справочнике принималось Гс-а - 1 77 и гс-вг 1 93 А. [40]
Однако для практических расчетов учет ядерных составляющих при вычислении термодинамических функций газов несуществен, так как химические равновесия шределяются изменениями термодинамических функций лри химических реакциях, а не их абсолютными значе-аиямн. [41]
Используя полученные выше соотношения, можно непосредственно перейти к вычислению термодинамических функций. [42]
Из рассмотрения данных этой таблицы следует, что при вычислении термодинамических функций СаНРСЬ по приближенным формулам ( XVIII. Приведенные в табл. 167 значения ФкОЛ ( СаНРСЬ) группируются около значений, вычисленных по формуле ( XVIII. [43]
До появления электронных вычислительных машин рядом авторов были предложены методы вычисления термодинамических функций двухатомных газов по молекулярным данным, в которых приближенно учитывались ангармоничность колебаний, центробежное растяжение и колебательно-вращательное взаимодействие. [44]
В § 8, 9 и 10 были рассмотрены приближенные методы вычисления колебательно-вращательных составляющих термодинамических функций двухатомных газов, а также методы, позволяющие учитывать в приближенных расчетах существование молекул в возбужденных электронных состояниях. Поскольку все приближенные методы вычисления колебательно-вращательных составляющих, кроме метода Джиока и Оверстрита, отличаются только способом вычисления поправок, учитывающих отклонения молекул газа от модели жесткий ротатор - гармонический осциллятор, соотношения, полученные в § 8 - 10, могут быть приведены к общему виду. [45]
Страницы: 1 2 3 4