О-метильная группа
Cтраница 2
Производное opmo - толуидина 115 при действии Н2О2 в АсОН превращается непосредственно в индолин 116 с выходом 70 %, эпоксид присутствует лишь в следовых количествах. Образование единственного продукта циклизации объясняется наличием о-метильной группы, отталкивающей ацетильный заместитель по направлению к алкенильному фрагменту, тем самым приводя к частичному экранированию последнего, что уменьшает число вероятных направлений атаки связи СС объемной частицей вольфраматного реагента. [16]
Влияние о-метильной группы на бромдесилилирование и ионное бромирование ароматических соединений приблизительно одинаково ( происходит ускорение реакции соответственно в 81 5 и 76 раз), хотя стерическое влияние на эти реакции должно быть различным. Из этого следует, что оказываемый о-метильной группой стерический эффект незначителен. Это подтверждается также и тем, что ускоряющее влияние о-метильной группы сильнее, чем / г-метильной. [17]
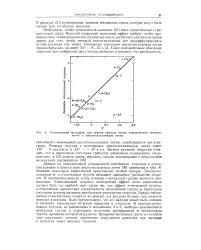 |
Гамметовский 0р - график для обмена протона между замещенными толуола-ми-а - и циклогексиламидом лития. [18] |
Влияние некоторых заместителей представляет особый интерес. Электроно-донорные м - и дг-алкильные группы вызывают ожидаемое уменьшение скорости. В противоположность этому влияние о-метильной группы является аномальным. Уменьшающий скорость электронный эффект этого заместителя должен быть по крайней мере таким же, как эффект л-метильной группы, а стерические препятствия копланарности метиленовой группы в переходном состоянии должны вызывать еще большее уменьшение скорости. [19]
В соединении Г две метальные группы в каждом бензольном цикле находятся в орго-положении по отношению к сульфонильной группе. Если принять, что вклад о-метильной группы в дипольный момент равен 0 3 D, то суммарный векторный вклад четырех метильных групп обоих бензольных циклов составит 0 6 D. Отсюда, расчетное значение дипольного момента соединения Г равно 5 87 - 0 6 5 27 D, что хорошо согласуется с найденным экспериментально. Таким образом, и эти данные подтверждают отсутствие требования копланарности для перекрывания орбиталей за счет Зс. [20]
Подобное влияние, однако, уже на стадии присоединения наблюдается при введении метильной группы в бензольное ядро бензилфосфоната. Так, диэтиловый эфир о-ксилилфосфоновой кислоты не реагирует с шиффовыми основаниями в эфирной среде при 10 - 15 С, тогда как эфиры га-ксилил-фосфоновых кислот реагируют гладко в этих условиях. Причиной этого различия также является пространственное влияние о-метильной группы, приводящее, по-видимому, к снижению нуклеофильности карбаниона, который можно считать изоэлектронным о-толуидину и не очень отличающимся от N-аниона продукта присоединения, полученного из бензилиден-о-толуиди-на и бензилфосфонового эфира. [21]
Влияние о-метильной группы на бромдесилилирование и ионное бромирование ароматических соединений приблизительно одинаково ( происходит ускорение реакции соответственно в 81 5 и 76 раз), хотя стерическое влияние на эти реакции должно быть различным. Из этого следует, что оказываемый о-метильной группой стерический эффект незначителен. Это подтверждается также и тем, что ускоряющее влияние о-метильной группы сильнее, чем / г-метильной. [22]
Важнейший из них ( схема 8) основан на использовании 5-грет-бутил - 1 3 4-тиадиазолил - 2-изоцианата, который с хорошим выходом получают действием фосгена на 2-амино - 5-трет-бутил - 1 3 4-тиадиазол. Продукт-реакции выделяют и кратковременно нагревают с хлороводородом для отщепления О-метильной группы. [23]
Реакция обмена толуолов, замещенных в кольцо, ускоряется электроноакцепторными и замедляется электронодонорными заместителями. Такая величина р значительно более положительна, чем для диссоциации фенолов в воде ( р 2 11); это показывает, что переходное состояние имеет в значительной степени карбанион-ный характер. Электронодонорные м - и и-алкильные группы, как и следовало ожидать, уменьшают скорость реакции, однако влияние о-метильной группы аномально. Полярный эффект этого заместителя должен быть по крайней мере таким же, как эффект тг-метильной группы [15], а пространственные препятствия достижению копланарности метиленовой группы с кольцом в переходном состоянии должны вызывать еще большее уменьшение скорости. [24]
Алкилирование арена повышает его основность и, следовательно, реакционную способность. Поэтому в лабораторных условиях обычно трудно остановить реакцию на нужной стадии алкилиро-вания. Так, из бензола, метилбромида и бромида алюминия образуется 1 2 4 5-тетраметилбензол, который более основен, чем низшие его алкильные гомологи, однако в дальнейшее алкилирова-ние это соединение вступает нелегко, так как оба свободных места в ядре блокированы соседними о-метильными группами, пространственно затрудняющими атаку электрофильного партнера. Для синтеза низших продуктов алкилирования следует выбирать мягкий катализатор Фриделя - Крафтса. [25]
Альдозы и кетозы обычно существуют в виде циклических по-луацеталей, поэтому при их взаимодействии с простыми спиртами в условиях кислотного катализа образуются полные ацетали, в которых сохраняется циклическая полуацетальная структура исходного сахара. Эта реакция, известная как синтез гликозидов по Фишеру [87], является термодинамически контролируемой и приводит к шестичленным пиранозидам. Причиной такого различия является то, что аномерный эффект ( см. разд. О-метильная группа может быть ква-за-аксиальной ( вследствие аномерного эффекта), а объемистая гидроксиметильная группа - ваза-экваториальной. Кроме того, в р-фуранозиде не должно быть вицинальных с-взаимодействий. Фуранозид ( 7 %) не может принимать такую предпочтительную конформацию, и у него 1 - О-метоксигруппа остается аксиальной. [26]
Так, в случае трифенилметильного радикала можно ожидать делокализацию радикального электрона по всем трем фениль-ным остаткам, в результате чего радикал стабилизируется, как изображено выше. Поскольку при этом идет речь об одном эффекте сопряжения, в полной мере взаимодействие возможно, однако, только тогда, когда три фенильных остатка лежат плашмя в одной и той же плоскости, так как иначе мезомерия пространственно затруднена ( см. также стр. Однако полностью плоскостное расположение в случае тритильного радикала не может осуществиться, так как три фенильных остатка мешают друг другу и поэтому принимают пропеллероподобное расположение. В три-о-толилметильном радикале поворот ароматических остатков из-за о-метильных групп в толильном остатке должен быть еще больше, поэтому объясняемая мезо-мерией стабилизация радикального электрона по сравнению с тритильным радикалом снижается. [27]
Возможен рост этих организмов в атмосфере СО с образованием ацетата. Большинство гомоацетогенов способно к росту на одноуглеродных субстратах. Acetobacterium woodii и некоторые клостридии в присутствии СО2 ( СО) используют О-метильные группы большого количества метоксилиро-ванных ароматических кислот, включая ванильную, сиринговую, 3 4 5-триметоксибензойную, ферулиновую и другие, для синтеза ацетата. [28]
Напротив, если в обоих орго-положениях бензольного ядра находятся объемистые заместители, индуктивным влиянием которых можно пренебречь, то пространственные препятствия делают невозможным копланарное расположение нитрогруппы и ароматического ядра, необходимое для заметного я, я-пере-крывания. Нитрогруппа оказывается в какой-то степени выведенной из плоскости кольца. Они изображены на рис. 12, в, где пространство, занятое о-метильными группами, схематически изображено окружностями. В результате пространственных препятствий между метальными группами и нитрогруппой ( заштрихованная поверхность) происходит поворот вокруг связи а в обозначенном стрелкой направлении. [29]
Стерические факторы, столь сильно влияющие на электронные спектры замещенных дифенилов, имеют, естественно, ту же природу, что и факторы, обусловливающие давно известное явление оптической изомерии в указанных системах ( см. Adams, Yuan, 1933; Adams, Finger, 1939); понимание этой общности оказывает существенную помощь при объяснении спектральной картины. Однако, отвлекаясь от условий возникновения асимметрии, следует отметить, что существуют две причины, вследствие которых электронные спектры являются гораздо более чувствительным индикатором заторможенного вращения, нежели наличие оптической активности. Во-вторых, оптическая активность зависит только от основного состояния молекулы, тогда как спектральная картина обусловлена основными и возбужденными электронными уровнями, а последние особенно подвержены влиянию стерических факторов. По этой причине; для появления оптической активности требуется значительно более высокая степень пространственного перекрывания, чем для выявления стерических эффектов в спектрах. Так, например, моно - и ди-ор / яо-замещенные дифенильг могут проявлять оптическую активность лишь при наличии очень крупных заместителей, тогда как даже наличие о-метильных групп очень сильно меняет спектральную картину. [30]
Страницы: 1 2 3