Генетический дефект
Cтраница 2
Таким образом, генетический дефект при болезни Вильсона - Коновалова ведет к нарушению выделения меди печенью при почти нормальном ее поступлении. Неспособность печени к нормальному выделению меди с желчью достоверно установлена и в опытах С. Т. Walsh ( 1985), показавших, что при этой болезни способность гепатоц итов выделять 1медь снижена по сравнению со здоровыми лицами в десятки и сотни раз. Сканирование проводят в положении лежа на спине от головы к ногам. На сканопрам ме здоровых лиц через 24 ч после внутривенного введения радиоизотопа в области печени появляется четкий пик, который через 48 и 72 ч дополняется вторым ликом в нижней части живота. У больных второй пик как при поступлении, так и после лечения хелати-рующими препаратами выражен очень слабо. [16]
Болезнь Вильсона-Коновалова - генетический дефект синтеза церулоплазмина: Гипераминоацидурия, снижение концентрации церулоплазмина, отложение меди в печени, почках, мозгу. [17]
Известен и ряд других генетических дефектов обмена меди у человека. [18]
Как уже отмечалось, многие генетические дефекты обмена меди у животных обнаруживают близкое сходство с наследственными заболеваниями у человека и могут рассматриваться как их модели. Это в первую очередь относится к наследственным дефектам обмена меди у мышей и собак. [19]
Параметр Т учитывает убывание частоты генетических дефектов в последующих поколениях. [20]
Клетки селезенки не обладают таким генетическим дефектом и поэтому миеломные клетки, не синтезирующие нуклеотиды, через определенноеиремя исчезают из культуры. Гибридомы остаются единственными делящимися клетками. Среду сменяют каждые сутки в течение двух дней, а затем - один раз через 48 часов. Примерно через 2 недели гибридомы начинают активно расти. [21]
Миодистрофия - группа нарушений, вызванных определенным генетическим дефектом и ведущих к деградации мышечной ткани и прогрессирующей мышечной слабости; пока не излечима. [22]
Ксантинурия - недостаточность ксантиноксидазы, вызванная либо генетическим дефектом, либо тяжелым поражением печени, приводит к гипоурикемии и увеличению экскреции оксипуринов - гипоксантина и ксантина. При тяжелой недостаточности ксантиноксидазы у пациентов часто развивается ксантинурия и образование ксантиновых камней. [23]
Две фундаментальные цели генной инженерии заключаются в исправлении генетических дефектов, таких как серповидно-клеточная анемия ( точечная мутация в гемоглобине), и в добавлении нормальных генов к другим, например включение гена нитроге-назы в хромосомы пшеницы. [24]

ДНК) были предприняты попытки использовать их для коррекции генетических дефектов. [26]
На основе указанных методов оценивают частоту индукции определенных типов генетических дефектов у различных поколений потомков и при равновесии. Равновесная частота устанавливается, если все последующие поколения потомков облучаются одинаковой дозой. В табл. 1 приведены ожидаемые частоты индукции генетических повреждений, рекомендуемые различными научными группами. При этом вся доза считается генетически значимой. [27]
Для здоровых людей тирозин является заменимой аминокислотой, но дети с генетическим дефектом, затрагивающим фенилаланин-ги-дроксилазу, для нормального роста должны получать тирозин с пищей. [28]
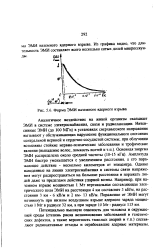 |
Форма ЭМИ наземного ядерного взрыва. [29] |
Потенциально высокую опасность для человека и окружающей среды ( степень риска возникновения заболеваний и генетических дефектов, а также вероятность тяжелых аварий и т.п.) составляют радиоактивные отходы и отработавшие ядерные материалы. [30]
Страницы: 1 2 3 4