Кинетика - суммарный процесс
Cтраница 2
Среди химических стадий, являющихся звеньями электрохимических процессов, особое значение имеют реакции с участием ионов Н и ОН -, определяющие зависимость кинетики суммарного процесса от рН раствора. Еще сравнительно недавно предполагалось, что в околоэлектродном слое кислотно-основное равновесие устанавливается со скоростью, которая велика по сравнению со скоростью процесса в целом. [16]
Практически, однако, при адсорбции органических веществ на s, р-металлах непосредственно адсорбционная стадия протекает значительно быстрее диффузионной, а потому не сказывается на кинетике суммарного процесса. Если же наблюдаемые 6, - кривые отклоняются от уравнения Корыта, то это, как правило, связано с замедленной перестройкой адсорбционного слоя. [17]
Характер взаимодействия полимерных тел с газообразными и жидкими средами в значительной степени определяется сорбцион-ными и диффузионными процессами, которые во многих случаях являются определяющими при оценке кинетики суммарных процессов взаимодействия. [18]
Это явление ппиписали многоступенчатому протеканию реакции, причем первой наиболее медленной стадией является взаимодействие между алкыгом ц катализатором, кинетика которого зависит от их концентраций, в то время как последующи стадии, по предположению, являющиеся весьма быстрыми, не оказывают влияния на кинетику суммарного процесса, протекание которого с кинетической точки зрения определяется наиболее медленной стадией. [19]
Вследствие подобного рода реакций становится возможным медленное нарастание скорости суммарного процесса со временем. С точки зрения кинетики суммарного процесса реакции VI и Via эквивалентны, так как в каждой из них малоактивный перекисныи радикал рождает активную частицу, продолжающую цепь реакции. Выбор между ними может быть сделан только на основании прямого анализа реагирующей смеси на содержание перекиси водорода. [20]
Таким образом, седлообразная неустойчивость представляет собой просто тепловой взрыв, определяемый кинетикой первой стадии реакции. Хотя тепло выделяется во второй стадии, но кинетика суммарного процесса при квазистационарной концентрации промежуточного продукта всецело определяется первой стадией. [21]
Согласно приведенной схеме, первая стадия практически равновесна, а вторая ( лимитирующая) - необратима. Последнее означает, что природа третьей стадии несущественна для кинетики суммарного процесса и, кроме того, в уравнении ( II 1.18) члены, содержащие W2, обращаются в нуль. [22]
Низкий порядок реакции по кислороду указывает на то, что стадия ( V) схемы, приведенной на стр. Положительный порядок реакции по бензальдегиду показывает, что стадия ( VI) влияет на кинетику суммарного процесса, или приводя к малой величине константы скорости, или же характеризуясь небольшой величиной константы равновесия. [23]
Таким образом, вопрос о влиянии хлоридов требует уточнения. Поскольку в смесителе происходит не только разложение NH4C1, но и частичная отгонка NH3 ( реакция ( 2)) и поскольку реакция ( 1) не является лимитирующей в данном процессе, мы сочли целесообразным исследовать кинетику суммарного процесса. Для этого мы применили методику эксперимента, описанную в работе [6], немного упростив ее. [24]
Реакции электрофильного замещения в ароматическом ядре являются одним из важнейших классов реакций органической химии. Однако изучение констант скоростей элементарной стадии этих процессов упирается в серьезную трудность - одним из реагентов являются промежуточные, как правило, гипотетические частицы ( например, NO), концентрация которых неизвестна. Непосредственному измерению поддается лишь кинетика суммарного процесса, который является сложным. Поэтому данных об энергиях активации и стерических факторах элементарного акта электрофильного замещения не имеется. [25]
Другой особенностью индия является его способность образовывать в растворах наряду с устойчивыми трехвалентными ионами также неустойчивые одновалентные ионы. Поскольку собственно электрохимическая реакция разряда-ионизации индия, вообще говоря, может протекать как совокупность нескольких электрохимических стадий с образованием промежуточных низковалентных частиц, то изучение условий накопления ионов 1п в ходе электродного процесса ( например, зависимость х концентрации от потенциала индиевого электрода) может способствовать выяснению механизма этого процесса. В частности, сопоставление результатов, исследования кинетики суммарного процесса с одновременным анализом закономерностей накопления частиц одновалентного индия позволяет решить одну из актуальных и сложных задач кинетики многостадийных процессов - являются ли возникающие при электродном процессе низковалентные частицы промежуточным продуктом этого процесса или конечным продуктом побочной электрохимической реакции, протекающей параллельно с основным электродным процессом. [26]
Концентрация растворенного вещества близка к нулю. Наблюдаемые закономерности определяются кинетикой процесса растворения. Если в раствор переходят предварительно адсорбированные молекулы, то при описании кинетики суммарного процесса следует учитывать закономерности процессов адсорбции и десорбции. [27]
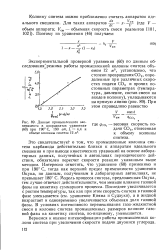 |
Данные промышленного эксперимента в координатах уравнения ( 60 при 190 С, 190 am, L 4 4 и объеме колонны синтеза 12 мя. [28] |
Это свидетельствует о том, что промышленные колонны синтеза карбамида действительно близки к аппаратам идеального смешения и при выводе кинетических уравнений на основе лабораторных данных, полученных в автоклавах периодического действия, обязателен пересчет скорости реакции указанным выше методом. Интересно отметить, что уравнение ( 60) применимо и при 190 С, тогда как верхний предел применимости уравнения Оцука, по данным, полученным в лабораторных автоклавах, не превышает 180 С. Модель процесса синтеза, предложенная Оцука, тем лучше отвечает действительности, чем меньше влияние газовой фазы на кинетику суммарного процесса. Последнее увеличивается с ростом температуры, так как при этом скорость синтеза в газовой фазе уменьшается ( см. уравнение Кияма и Судзуки), а в жидкой возрастает и одновременно увеличивается объемная доля газовой фазы. В условиях интенсивного перемешивания газо-жидкостной смеси в колонне синтеза промышленных размеров влияние газовой фазы на кинетику синтеза, по-видимому, уменьшается. [29]
Для самых быстрых стадий в этих условиях скорости прямой и обратной реакции практически равны. Концентрации молекул, участвующих в этих стадиях, или концентрации образующихся из них продуктов при малых заполнениях поверхности вовсе не появляются в кинетических уравнениях суммарного процесса, если эти стадии совершаются после медленной стадии, называемой контролирующей. Напротив, быстрые стадии, непосредственно предшествующие контролирующей, явно влияют на кинетику суммарного процесса, и равновесная концентрация одного из продуктов быстрой стадии появляется в наблюдаемом кинетическом уравнении. Часто вместо этих концентраций фигурируют тождественные им произведения из константы равновесия соответствующей быстрой стадии и вытекающего из ее стехиометрии произведения концентраций исходных веществ, участвующих в этой стадии. [30]
Страницы: 1 2 3