Истинная кинетика
Cтраница 2
Очевидно, что в диффузионной области наблюдаемая макроскопическая кинетика реакции не имеет ничего общего с истинной кинетикой на поверхности. Все реакции в диффузионной области имеют первый порядок по концентрации реагирующего вещества при постоянном общем давлении. При этом порядок реакции по общему давлению отличается от порядка по концентрации. [16]
Для получения необходимых данных о химической реакции при помощи соотношений Онзагера нужно учитывать следующее [29]: истинная кинетика химической реакции часто неизвестна и принцип частичного равновесия непосредственно применять нельзя. [17]
Область условий, при которых можно не учитывать диффузионные факторы, называется кинетической областью, или областью истинной кинетики. [18]
Однако точность зависимости этого критерия от параметров Ф5 или Фь в большой степени определяется надежностью данных об истинной кинетике, точностью определения эффективного коэффициента диффузии и изотермич-ностью гранулы. [19]
В заключение отметим, что если замедленной является какая-либо недиффузионная стадия, например переход заряда, то принято говорить об истинной кинетике процесса, а если диффузионная - то о диффузионной кинетике. Очень часто скорости стадии перехода заряда ( или химической реакции) и диффузионной соизмеримы. [20]
Известно, что для реакции углерода с кислородом легко наблюдалась чисто диффузионная область без малейшего следа каких-либо искажений, связанных с истинной кинетикой на поверхности. Это объясняется именно тем, что истинный порядок реакции ниже первого и может служить лишним доводом в пользу последнего заключения. [21]
Очевидно, что протекание реакции в условиях существенных ограничений массопередачи между газовыми пузырями и жидкостью эквивалентно случаю работы катализатора при определяющей роли истинной кинетики, но при пониженном давлении газа. Поэтому влияние этого вида ограничений массопереноса на селективность реакции аналогично влиянию концентрации водорода в жидкости на истинную селективность реакции. [22]
До настоящего времени основная информация о процессах переработки тяжелых нефтяных фракций пос - тупает с проточных интегральных реакторов, что не позволяет выявить истинную кинетику тех или иных превращений и вынуждает ограничиваться исследованиями процесса в целом при наличии отмеченных выше осложнений. [23]
Если при некоторых определенных условиях наиболее медленной стадией процесса является сама химическая реакция ( химическое взаимодействие), то скорость суммарного процесса определяется истинной кинетикой данной химической реакции. [24]
Если при некоторых определенных условиях наиболее медленной стадией процесса является сама химическая реакция ( химическое взаимодействие), то скорость суммарного процесса определяется истинной кинетикой данной химической реакции. Совокупность условий, при которых наблюдается такая закономерность протекания процесса, носит название кинетической области. [25]
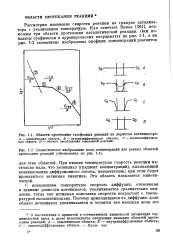
При низких температурах скорость реакции настолько мала, что потенциал ( градиент концентраций), вызывающий возникновение диффузионного потока, незначителен; при этом будет проявляться истинная кинетика. Эта область называется кинетической. [27]
Уже сравнительно давно было отмечено [1,3,8] t что отсутствие зависимости ( или слабая зависимость) стационарной скорости растворения пассивного металла от потенциала ни в коей мере не характеризует истинную кинетику самого процесса растворения. В этом случае влияние потенциала является более сложным, поскольку его рост приводит не только к обычному ускорению анодного растворения металла, но и к изменению состояния металлической поверхности, которое равноценно повышению перенапряжения того же процесса. По-видимому, в случае железа и хрома эти эффекты полностью компенсируют друг друга, что и приводит к независимости стационарной скорости растворения этих металлов в пассивном состоянии от потенциала. Поскольку, однако, характерное для каждой величины потенциала стационарное состояние поверхности устанавливается относительно медленно, эти два эффекта удается разделить, если применить метод быстрого наложения поляризации. [28]
Однако в отличие от Смита и Юэрта, которые рассматривали второй случай только как гипотетический для определения нижнего значения числа частиц, Гардон считал, что он описывает истинную кинетику первой стадии. [29]
Если же медленной стадией процесса является подвод реагентов к поверхности или отвод продуктов реакции, то скорость процесса всецело определяется скоростью диффузии, и макроскопическая кинетика реакции не имеет ничего общего с истинной кинетикой на поверхности. Эту предельную область гетерогенного процесса называют диффузионной. [30]
Страницы: 1 2 3 4