Атом-атом
Cтраница 1
Атом-атом потенциалы в принципе могут дать информацию как о фазовом состоянии вещества при определенных внешних условиях, так и о деталях взаимного расположения молекул. Однако далеко не все проблемы, касающиеся структуры полимерных кристаллов, могут быть решены расчетами с применением атом-атом потенциалов. Это объясняется тем, что, во-первых, не все молекулы полимера строго идентичны ( имеется некоторое распределение по молекулярным весам, а также вполне определенная степень тактичности), а, во-вторых, при кристаллизации часть молекул или отдельные их участки не успевают распутаться. Таким образом, кинетический фактор играет далеко не последнюю роль, и поэтому структура полимерного кристалла часто определяется условиями кристаллизации. [1]
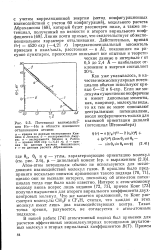 |
Потенциал взаимодейст взаимной ориентации диполей вия Не - - - Не в области взаимного ( потенциал Штокмайера отталкивания атомов. [2] |
Атом-атом потенциалы обычно не используются для исследования взаимодействий молекул газов. В прошлом было предпринято несколько попыток применения такого подхода [70, 71], однако они не вызвали интереса, поскольку об атом-атом потенциалах тогда еще было мало известно. Интерес к атом-атомному подходу вновь возрос лишь недавно [72, 73], причем Конг [73] получил выражения для второго вириального коэффициента двух-центровых молекул. Что же касается приложений, то Конг рассмотрел молекулы CH3F и CF H, считая, что каждая из этих молекул имеет лишь два взаимодействующих центра. Такая точка зрения, разумеется, не согласуется с атом-атомным подходом. [3]
 |
Энергия активации для реакций некоторых замещенных метана. [4] |
Атом-атом потенциалы являются более тонким инструментом исследования переходного состояния, чем рассмотренные выше аддитивные схемы. [5]
Методом атом-атом потенциалов была рассчитана равновесная геометрия производных изобутана, адамантана, гомоадамантана, норборнана и бицикло [2,2,2] октана IV - VIII в основном и переходном состояниях и определена их энергия напряжения. [6]
Метод атом-атом потенциалов позволяет понять некоторые интересные особенности упаковки макромолекул в кристаллах. [7]
Самые простые атом-атом потенциалы, как уже указывалось, это стенки бесконечной высоты ( рис. 2.1 а), соответствующие всего лишь одному эмпирическому параметру гт. Если г гт, то контакты между атомами разрешены, в противном же случае они запрещены. Такого рода потенциалы, соответствующие методу жестких сфер в конформационном анализе или принципу плотной упаковки молекул в кристаллах, дают информацию о разрешенных и запрещенных областях в пространстве параметров, описывающих геометрию молекулы. Надо сказать, что метод жестких сфер, вообще говоря, весьма мало дает для малых перегруженных молекул, не обладающих внутренним вращением, однако для таких молекул, как пептиды, в которых конформа-ционная свобода относительно высока, он дает возможность объяснить некоторые интересные факты. [8]
Определение параметров атом-атом потенциалов по всей совокупности физико-химических данных с использованием четкого математического критерия становится по изложенным причинам весьма важной задачей. Универсальные потенциалы должны использоваться для вычисления таких разных свойств, как параметры элементарной ячейки кристалла, теплота сублимации, термодинамические функции кристалла, термохимические свойства газов, конформаций молекул, частоты колебательных спектров изолированных молекул и кристаллов, второй вириальный коэффициент и свойства переноса многоатомных газов, данные по рассеянию молекулярных пучков. Но каковы должны быть оптимальные потенциалы. [9]
Другие известные потенциалы атом-атом взаимодействий уже не столь хороши, как указанные выше, поскольку они содержат большее число эмпирических параметров. [10]
Итак, схема атом-атом потенциалов оказалась вполне приемлемой для расчетов термохимических параметров таких разных систем, как алканы, олефины, ароматические молекулы, цикло-алканы и циклоалкены, бициклические и полициклические системы и, наконец, галогенпроизводные. Насколько же оправдан такой подход к расчетам энтальпий атомизации более сложных молекул, как, например, кислородсодержащих молекул или нитро-соединений, без детального анализа сказать трудно, однако тот факт, что грубые аддитивные схемы недурно работают для таких сложных систем, позволяет надеяться и на успех схемы атом-атом потенциалов. Преимущество атом-атомного подхода очевидно - он является ключом к пониманию связи между геометрией молекул и термохимическими свойствами веществ. [11]
Таким образом, как атом-атом потенциалы, так и более грубый критерий допустимых контактов, дают очень много для понимания конформации ди - и полисахаридов а в некоторых случаях они просто необходимы при интерпретации рентгеноструктурного эксперимента. Внутримолекулярные взаимодействия ( ван-дер-ваальсовы и водородные связи) играют первостепенную роль в определении оптимальных конформации, однако в тех случаях, когда возможны межцепные водородные связи, приводящие либо к многотяжевым спиралям, либо к различным полиморфным модификациям, имеет смысл учитывать и межмолекулярные взаимодействия. [12]
В основе применения метода атом-атом потенциалов к молекулам лежит предположение, что внутримолекулярные взаимодействия носят тот же характер, что и межмолекулярные. Трудно сказать, насколько это предположение верно. Косвенным его оправданием является целый ряд экспериментальных и теоретических фактов. Можно еще отметить, что методы молекулярных орбиталей дают обычно лучшие результаты, если в качестве базисных рядов используются атомные орбитали, а не какие-нибудь другие. Словом, атомы, входя в состав молекул, сохраняют многие свои свойства. [13]
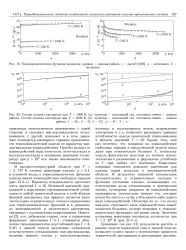
Причем вклады от взаимодействий вида атом-атом, атом-молекула и молекул а-молекул а в указанном диапазоне температур при р Ю3 атм также оказываются сопоставимы. [15]
Страницы: 1 2 3 4