Атом-атом
Cтраница 3
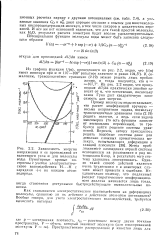
Как сказываются электростатические взаимодействия на деформациях молекулы, сравнимо ли их действие с действием атом-атом потенциалов. [32]
Ввести в потенциальную функцию член, зависящий не только от расстояний между ядрами ( сумма атом-атом взаимодействий), но и член, зависящий от взаимного расположения векторов, соединяющих ядра, точнее от угла между группами определенных векторов. [33]
Следует сказать, что энтальпии атомизации ( образования, изомеризации) весьма чувствительны к параметрам атом-атом потенциалов. Если для равновесных конформаций существенна скорость изменения отталкивания атомов с увеличением расстояния между ними ( действительно, условие равновесия определяется обращением в нуль первых производных), то на термохимических свойствах больше сказываются абсолютные значения энергии. Поэтому неудивительно, что атом-атом потенциалы, удовлетворительно предсказывающие структуру молекул, могут давать не согласующиеся с опытом термохимические оценки. [34]
Рассматривая мономерные единицы или нуклеотидные фрагменты, содержащие не более одного основания, мы могли ограничиться учетом только атом-атом потенциалов. При переходе к олиго-мерам и полимерам важно принять во внимание большую роль гидрофобных, точнее, сольватофобных взаимодействий. Действительно, нативные нуклеиновые кислоты, в которых основания расположены стопкообразно, существуют только в водных растворах в определенном интервале температур; в органических растворителях или при повышении температуры они денатурируют. [35]
Основам конформационного анализа малых молекул посвящены первые шесть глав, и лишь в трех последних главах изложено применение модели атом-атом потенциалов к макромолекулам - синтетическим полимерам, полисахаридам, белкам и полинуклеотидам. Многие сочтут, вероятно, что последние главы следовало бы написать более фундаментально, как, собственно, и было задумано вначале. Но это должно было бы привести к значительному увеличению объема рукописи. Остается лишь втайне надеяться, что когда-нибудь мне посчастливится написать еще одну монографию, специально посвященную конфор-мациям макромолекул. [36]
Кроме статистического подхода, для тех же целей нахождения спиральных и неспиральных участков может быть применен подход, основанный на атом-атом потенциалах. Для этого естественно рассмотреть взаимодействия ближайших структурных единиц: эти взаимодействия проявляются либо в дипептидах, либо в других фрагментах, из которых строятся белки. [37]
Синтетические регулярные макромолекулы, на конформациях которых мы вначале остановимся, представляют собой очень удобные объекты для проверки эффективности метода атом-атом потенциалов. В последующем изложении основное внимание уделено кристаллическим макромолекулам, поскольку их потенциальные функции достаточно просты и зависят лишь от нескольких переменных. Для макромолекул в растворах, разумеется, невозможно получить столь же детальное описание геометрии, однако в этом случае имеет смысл говорить о средних характеристиках. [38]
К сожалению, теплота сублимации полиацетальдегида неизвестна, и мы не можем сказать, в какой степени удовлетворительна глубина потенциальных ям в атом-атом потенциалах. [39]
Используя это поле в расчетах, обычно находят силовые постоянные из некоторой совокупности экспериментальных данных, и тогда проблема переносимости потенциалов для атом-атом взаимодействий заменяется проблемой изменения постоянных F tj и Fti при переходе от одной молекулы к другой. Далее, не вполне ясен смысл членов, линейных относительно изменений длин связей, валентных углов и расстояний между несвязанными атомами. В равновесной конформации эти члены должны были бы обратиться в нуль, если бы между параметрами, используемыми в выражении (4.27), не было зависимости. Пдэтому представляется более последовательным проводить расчеты частот колебаний молекул в общем гармоническом силовом поле, не включая линейных членов. Для этого зависимые геометрические параметры, входящие в выражение для потенциальной функции, должны быть выражены через независимые, и тогда матрица силовых коэффициентов может быть найдена по уравнениям (4.25) однозначно для любого выбранного набора независимых параметров. [40]
При расчете коэффициентов переноса необходимо определить достоверные потенциальные кривые для взаимодействия атомов и молекул щелочных металлов, причем особенно важно знать энергию взаимодействий атом-атом и атом-молекула, так как именно эти взаимодействия вносят основной вклад в коэффициенты переноса. [41]
Но не следует думать, что неэмпирические расчеты могут полностью заменить полуэмпирические методы квантовой химии, а также слабо обоснованные эмпирические методы, использующие атом-атом потенциалы. Большинство органических молекул, интересующих химиков, устроено слишком сложно, для того чтобы их геометрию и конформационные энергии можно было бы рассчитать, решая каждый раз хартри-фоковские задачи. Трудно представить и возможность минимизации хартри-фоковского функционала по независимым геометрическим параметрам: для молекул средних размеров такая процедура потребовала бы неразумно много машинного времени. Поэтому можно ожидать, что эмпирические методы первыми будут применяться к новым и неожиданным задачам, подобно тому, как атом-атом потенциалам всегда предшествует анализ пространственных моделей. [42]
Конечно, отталкивание аксиального атома в этих соединениях невелико ( порядка нескольких сотых или десятых ккал / моль), но если исходить из модели атом-атом потенциалов, то во всех случаях равновесие должно было бы сдвигаться в сторону экваториальных конформеров. [43]
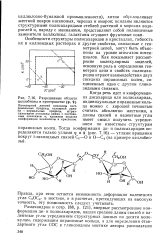 |
Разрешенные области целлобиозы в пространстве ( р, ф. [44] |
Особенности структуры полисахаридов в кристаллах, гибкость их в коллоидных растворах и другие свойства, связанные с геометрией цепей, могут быть объяснены на уровне атом-атом потенциалов. [45]
Страницы: 1 2 3 4