Метод - попло
Cтраница 3
Интегралы кинетической энергии предлагается оценивать непосредственно используя ОСЦ. Но такой подход вызывает очевидные возражения, во всяком случае в рамках метода Попла. Это относится особенно к резонансным интегралам между несвязанными атомами, а ОСЦ являются очень плохим приближением к АО Хартри - Фока в областях, удаленных от ядер. Даже аналитические приближения к АО Хартри - Фока, получаемые по методу Рутана, оказались мало удовлетворительными в этих областях, поскольку АО этого типа выбираются на основании требования минимизации полной энергии рассматриваемого атома, а эта величина определяется частями АО, близкими к ядру. [31]
Такая аналогия заставляет обратить внимание на одно важное обстоятельство. Разумеется, метод ВМО является гораздо более приближенным, чем метод Попла; однако он имеет преимущество: приводит непосредственно к важным теоремам, устанавливающим соотношения между свойствами и строением молекулы. Метод Попла позволяет вычислять свойства только отдельных молекул. Все попытки делать общие выводы на основании таких расчетов аналогичны попыткам прийти к каким-то эмпирическим заключениям на основании экспериментальных данных. Короче говоря, метод Попла может в лучшем случае заменить эксперимент, однако с помощью этого метода нельзя вывести общие принципы, которые необходимы химикам в их повседневной работе. Метод Попла не является точным методом, тем не менее даже если бы мы умели находить точные решения уравнения Шредингера для больших молекул, это все равно не дало бы нам того, в чем мы нуждаемся. Для того чтобы можно было планировать синтез, делать какие-то заключения о механизмах реакций или определять, структуру молекул, необходимы некоторые общие представления, которыми мы могли бы руководствоваться. [32]
Иногда подразделяют метод Пари. Под методом Попла понимают уравнения Рутаана в НДП и л-электронпом приближении. Метод Паризера - Парра является методом конфигурационного взаимодействия. [33]
Эта модификация ненамного сложнее обычного метода МОХ, и соответствующие уравнения (3.86) и (3.87) легко можно решить с помощью вычислительных машин. Однако приведенные в предыдущем разделе соображения показывают, что это довольно бессмысленное. Включение перекрывания в метод Попла не дает никаких преимуществ, если используется полуэмпирический подход. Это должно быть еще более справедливо для гораздо более грубого метода МОХ: учет перекрывания в нем является совершенно бесполезным уточнением. Действительно, имеющиеся данные подтверждают, во-первых, что включение перекрывания не приводит ни к каким практическим преимуществам, во-вторых, что модифицированный таким образом метод по-прежнему непригоден для количественных расчетов, и, в-третьих, что в некоторых случаях учет перекрывания вообще только ухудшает положение ( см. гл. [34]
Попла [16, 21], оптимально сочетающий логическую схему метода самосогласованных ЛКАО - МО Рутана и малую трудоемкость полуэмпирического метода Хюккеля. Напомним, что метод Попла фактически представляет собой переформулировку метода Рутана в приближении нулевого дифференциального перекрывания ( НДП), при котором пренебрегают перекрыванием орбит разных атомов и, как следствие этого, из всех двухзлектронных интегралов учитывают только интегралы кулоновского типа. Для наших целей метод Попла привлекателен тем, что в нем я-электронная система кумулена рассматривается как единое целое. Это позволяет выяснить ряд ее специфических особенностей, в частности взаимное влияние двух ортогональных систем сопряженных л-связей. [35]
 |
Влияние вращения осей координат на уравнения Попла для. [36] |
Повернем оси координат на угол Э ( рис. 3.3 6) и исследуем уравнения Попла, с тем чтобы выяснить, приведет ли такое вращение к каким-либо изменениям и если приведет, то к каким именно. Если уравнения значительно изменятся, это будет означать, что результаты метода Попла не инвариантны относительно поворота осей координат. [37]
Изложенный выше первоначальный вариант метода Попла логически неудовлетворителен в двух отношениях. Во-первых, АО различных атомов не подчиняются условию (3.25); это относится и к любому другому ряду функций, которые могли бы служить базисным набором в методе Рутана. Во-вторых, как мы уже отметили, из выражения (3.25) следует, что остовные интегралы НСц ( Pi) должны быть равны нулю; однако если ввести это предположение, то метод Попла приводит к неудовлетворительным результатам. В этом разделе мы проанализируем метод Попла более подробно и посмотрим, нельзя ли что-нибудь сделать для его улучшения. [38]
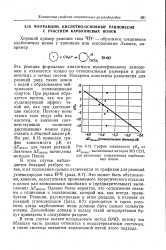 |
График зависимости рКА от Д. дел ( ж, вычисленных методом МО ССП, для различных сопряженных карбониевых ионов. [39] |
Эта реакция формально аналогична нуклеофильному замещению и отличается только по относительным размерам и роли нечетных и четных систем. Измерены константы равновесия для реакций ряда таких ионов с водой с образованием нейтральных ненасыщенных спиртов. На рис. 8.13 приведен график зависимости рК от Доделок для таких реакций. Значения Доделок вычислены методом Попла. [40]
Изложенный выше первоначальный вариант метода Попла логически неудовлетворителен в двух отношениях. Во-первых, АО различных атомов не подчиняются условию (3.25); это относится и к любому другому ряду функций, которые могли бы служить базисным набором в методе Рутана. Во-вторых, как мы уже отметили, из выражения (3.25) следует, что остовные интегралы НСц ( Pi) должны быть равны нулю; однако если ввести это предположение, то метод Попла приводит к неудовлетворительным результатам. В этом разделе мы проанализируем метод Попла более подробно и посмотрим, нельзя ли что-нибудь сделать для его улучшения. [41]
Первые расчеты такого рода [11, 12, 14] ограничивались ароматическими углеводородами, в которых все связи С-С сходны друг с другом. Поэтому предполагалось, что энергии ст-связей также должны быть примерно одинаковыми, так что для энергии ароматической а-связи можно использовать некоторое общее значение. Теплоты атомизации были вычислены как методом Попла, так и методом РПО, причем в обоих случаях результаты очень хорошо согласовывались с экспериментом. [42]
Основное достоинство этого метода состоит в том, что мы можем найти орбитальные коэффициенты и энергии МО непосредственно, не прибегая к процедуре самосогласования. Следует отметить, что когда длины связей в молекуле равны, а распределение электронной плотности однородно, хюкке-левские МО совпадают с самосогласованными. Поэтому при рассмотрении молекул, близко удовлетворяющих этим требованиям, часто бывает достаточным ограничиться подстановкой в уравнения Попла (1.5) - (1.7) хюккелевских коэффициентов. Иными словами, в обычном варианте метода Попла собственные векторы аппроксимируются хюккелев-скими ЛКАО - МО. [43]
Такое рассмотрение полностью самосогласовано, так как энергия сг-связей вычисляются непосредственно как фунции длин связей из предполагаемого значения для чистой а-связи и соответствующей силовой постоянной. Еще одной интересной деталью является тот факт, что самосогласованность такого рода достигается только в методе Попла. По-видимому, лишняя поправка на корреляцию в подходе РПО не нужна, а метод Попла включает достаточно параметров для того, чтобы удовлетворительно учесть все виды корреляции. Такой вывод тоже был весьма приятен, особенно если учесть, что и метод РПО требует введения дополнительных параметров. [44]
Такая аналогия заставляет обратить внимание на одно важное обстоятельство. Разумеется, метод ВМО является гораздо более приближенным, чем метод Попла; однако он имеет преимущество: приводит непосредственно к важным теоремам, устанавливающим соотношения между свойствами и строением молекулы. Метод Попла позволяет вычислять свойства только отдельных молекул. Все попытки делать общие выводы на основании таких расчетов аналогичны попыткам прийти к каким-то эмпирическим заключениям на основании экспериментальных данных. Короче говоря, метод Попла может в лучшем случае заменить эксперимент, однако с помощью этого метода нельзя вывести общие принципы, которые необходимы химикам в их повседневной работе. Метод Попла не является точным методом, тем не менее даже если бы мы умели находить точные решения уравнения Шредингера для больших молекул, это все равно не дало бы нам того, в чем мы нуждаемся. Для того чтобы можно было планировать синтез, делать какие-то заключения о механизмах реакций или определять, структуру молекул, необходимы некоторые общие представления, которыми мы могли бы руководствоваться. [45]
Страницы: 1 2 3 4