Нуклео-фил
Cтраница 2
Прибавление в процессе сольволиза к реакционной смеси более сильных нуклео-филов, чем растворитель, должно было бы оказывать гораздо меньший эффект на скорость сольволиза, происходящего по предельному S 1 механизму, чем на скорость сольволиза по SN2 механизму. Действительно, любой нуклеофил, который может конкурировать с растворителем в бимолекулярном процессе, будет ускорять реакцию, в то время как состояние промежуточного карбониевого иона, образующегося в мономолекулярной реакции, не сказывается на уравнении скорости. Влияние щелочных добавок на скорость аллильных сольнолктических реакций находится в соответствии с заключениями о механизмах, сделанными на основе изучения эффектов растворителя. Этот критерий механизма применяли также Де ля Map и Верной [45, 49], которые нашли, что скорости реакции хлористого аллила, хлористого р-метилаллила и 1 3-дихлорпропена с этанолом и водным этанолом довольно чувствительны к добавлению щелочи, тогда как на сольволиз хлористых а а - и у у-диметилаллилов щелочь не оказывает влияния. Сольволиз хлористого циннамила и хлористого а-метилаллила в очень незначительной степени подвержен влиянию щелочной добавки, в то время как сольволиз хлористого кротила в отношении чувствительности к основанию находится между сольволизом хлористого аллила и хлористого метияаллила. Заключения о механизмах, сделанные на основе этих наблюдений, находятся в полном соответствии с выводами, базирующимися на эффектах растворителей. [16]
Относительная реакционная способность катионов по отношению к стандартному нуклео-филу ( анизол) изменяется в ряду: хиназолиний пиримиди-ний 5-метилпиримидиний 2-аминопиримидиний 4-метил-пиримидиний 4-фенилпиримидиний 2 4-диметилпиримиди-ний. [17]
Анион F - не сольватирован и действует как активный нуклео-фил. [18]
Скорость реакции не зависит от концентрации и природы нуклео-фила, так как он не участвует в стадии, определяющей скорость реакции. [19]
В то же время различие между реакционной способностью нуклео-филов в газовой фазе значительно меньше, чем в растворе. Это, связано с тем, что реакции в газовой фазе идут с очень большими скоростями. Таким образом, еще раз подтверждается вывод о том, что очень большой вклад в энергию активации реакций бимолекулярного замещения вносит энергия десольватации реагентов. [20]
Известно много примеров того, что если субстрат и нуклео-фил не взаимодействуют самопроизвольно, они могут начать реагировать при стимулировании ближним УФ-светом. Природа стадии инициирования и действительный механизм фотоиници-ированных процессов до сих пор не ясны. Более того, многие фотоинициированные реакции могут включать цепные и нецепные процессы, протекающие конкурентно. [21]
Реагент Y: или растворитель может действовать и как нуклео-фил по отношению к атому углерода, и как основание по отношению к атому водорода при соседнем углеродном атоме. Поэтому реакции нуклеофильного замещения и элиминирования конкурируют друг с другом. В мономолекулярном процессе Е - механизм конкурирует с ЗдгЬмеханизмом замещения. BNl и заключается в ионизации субстрата с образованием карбокатиона. Во второй стадии El - процесса растворитель отщепляет протон от р-углеродного атома, тогда как при Sjyl-механизме происходит атака растворителя по положительно заряженному атому углерода. Это составляет единственное различие двух родственных реакций - отщепления и замещения. Как уже было отмечено ранее, для карбокатионов скорость захвата нуклеофильного агента превышает скорость депротонирования, т.е. S l El, и это соотношение практически не удается изменить заменой растворителя или нуклеофильного агента. Изменение в соотношении продуктов 8 1-замещения и Е1 - эли-минирования может быть достигнуто в ионизирующих, полярных и ненуклеофильных растворителях, таких, как трифторме-тансульфокислота, и суперкислотах FSO3H - SbF5, SO2C1F - SbF5 или в тех случаях, когда карбокатионный центр стерически полностью блокирован разветвленными алкильными группами. [22]
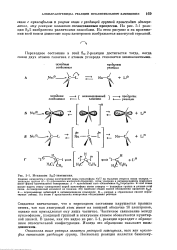 |
Механизм 8 2-замещения. [23] |
Описанная выше реакция является реакцией замещения, так как нуклео-фил вытесняет уходящую группу. [24]
На первой стадии реакции анион - CN атакует как нуклео-фил атом углерода карбонильной группы. [25]
Первичные и вторичные амиды, так же как и другие нуклео-филы, в кислых, нейтральных или основных растворах присоединяются к карбонильной группе альдегидов и кетонов. Первоначальным продуктом обычно является N-ацилкарбиноламин схема ( 144), который образуется в результате замещения у амидного азота. В отличие от простого алкили-рования N-замещенные продукты образуются даже в нейтральной среде, вероятно, в связи с тем, что перегруппировка имидата в амид облегчается в присутствии гидроксиметиленового заместителя. Температура ( 100 - 150 С), необходимая для проведения реакции, также облегчает O - N-перегруппировку. Реакция легче протекает, если кетоны содержат электронооття-гивающие а-заместители ( например, гексафторацетон, ацилоины или а-аминоалкилкетоны), хотя в двух последних случаях происходит последующая конденсация. [26]
В противоположность этому, реакции SN2, которые требуют участия сильных нуклео-филов ( обычно они представляют собой сильные основания), чаще всего протекают в нейтральных или основных условиях. Другое обстоятельство, которое усиливает нуклеофугные свойства уходящих групп, это напряженность цикла. Непрото-нированные простые эфиры не расщепляются совсем, а прото-нированные - только в жестких условиях; в то же врямя эпо-ксиды [289] расщепляются довольно легко, а протонированные эпоксиды - еще легче. [27]
Строение продуктов присоединения R0 - и Gp - показывает, что нуклео-фил атакует циклобутадиеновое кольцо с внешней стороны относительно центрального атома металла. [28]
 |
Дополнительные литературные данные о получении сложных эфиров. [29] |
В обычных условиях замещения в протонных растворителях карбоксилат-анион является одним из наиболее слабых нуклео-филов. Главный фактор, снижающий нуклеофильность, - сильная сольватация аниона. [30]
Страницы: 1 2 3 4