Подвижность - полимерная цепь
Cтраница 1
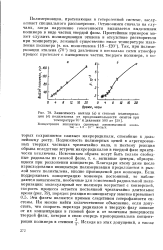 |
Зависимость выхода ( а и степени полимеризации ( б полиэтилена от продолжительности опытов при температуре 70 и давлении 500 ат. [1] |
Подвижность полимерных цепей в агрегированных твердых частицах чрезвычайно мала, и поэтому реакция обрыва вследствие встречи макрорадикалов в твердой фазе практически исключена. Благодаря этому даже после израсходования инициатора полимеризация продолжается в рыхлой массе полиэтилена, вполне проницаемой для мономера. Если поддерживать концентрацию мономера постоянной, то наблюдаются зависимости, необычные для гомогенной радикальной полимеризации: молекулярный вес полимера возрастает с конверсией, скорость процесса остается постоянной чрезвычайно длительное время ( рис. 79), порядок реакции по инициатору заметно превышает 0.5. Эти факты являются прямым следствием гетерофазности системы. [2]
Рост подвижности полимерной цепи при увлажнении объясняется адсорбцией молекул воды и их взаимодействием, например с гидрокеильными группами, участвующими в образовании межмолекулярных водородных связей в эпоксидных, фенольных и других клеях. Кажущаяся энергия активации [87] разрушения эпоксидной смолы, отвержденной аминными отвердителями в атмосфере со 100 % - ной влажностью, составляет от / з до / г энергии активации разрушения в сухом состоянии. [3]
 |
Свободная ( а п деформированная ( б полимерные сетки. вытягивание цепей для наглядности сильно преувеличено. [4] |
Типичным механизмом подвижности полимерных цепей в концентриров. [5]

В стеклообразном состоянии подвижность полимерной цепи отсутствует, во всех остальных состояниях она имеется. Исключение составляют лишь жесткоцепные системы. [7]
В высокоэластическом состоянии подвижность полимерных цепей велика, и это определяет общие закономерности и природу трения высокоэластических полимеров. Здесь ярко выражена зависимость силы трения от скорости скольжения, температуры и давления. Теоретической основой и интерпретацией этих закономерностей являются молекулярно-кинетические представления о подвижности молекул полимера на границе контакта с твердым телом. Разработанная на основе этих представлений общая теория трения высокоэластических полимеров позволила не только объяснить ряд важных экспериментальных зависимостей, но и предсказать новые. [8]
Однако данные, характеризующие подвижность полимерной цепи, можно получить, определяя скорость фотохромного процесса. [9]
 |
Зависимость величины смещения температуры максимума tg A от продолжительности отверждения. [10] |
Твердая поверхность, ограничивая подвижность полимерных цепей, сказывается на кинетике сшивания макромолекул. [11]
При понижении температуры уменьшается подвижность полимерных цепей, что приводит к уменьшению эластичности каучуков и резин. [12]
 |
Схематическое изображение изменения температурной зависимости деформации жесткоцепного полимера при введении пластификатора. [13] |
В самом деле, степень подвижности полимерных цепей определяется для гибких полимерных молекул значением времен релаксации их при наложении деформирующих усилий. [14]
Взаимодействие с подложкой приводит к ограничению подвижности полимерных цепей, что эквивалентно образованию дополнительных узлов полимерной сетки. Однако за счет изменения структуры в поверхностном слое и снижения плотности упаковки в нем общая плотность пограничного слоя может снижаться. Таким образом, интегральный эффект зависит от вклада каждого из этих процессов. О конечном результате можно судить, например, по сравнительным данным о набухании полимера, его температуре стеклования, плотности и др. Наиболее распространены подобные исследования для наполненных полимеров. Данные для наполненных систем можно распространить на поведение полимера в адгезионных соединениях, разумеется, с известными ограничениями, связанными с тем, что, например, в клеевом шве полимерная прослойка не может быть столь тонкой, как в сильно наполненном полимере. [15]
Страницы: 1 2 3 4