Гибкие полимер
Cтраница 3
Приведенные здесь данные получены на основе приближенных уравнений, и сами расчеты применимы лишь для частного вида цепей. Тем не менее, эти данные могут рассматриваться как типичные для гибких полимеров. Они будут очень полезны для конкретного рассмотрения термодинамических и гидродинамических свойств таких молекул в последующих главах. [31]
В то же время, как показывает опыт, согласие отсутствует для таких полимеров, как производные целлюлозы, дезоксирибонуклеиновая кислота и некоторые другие. Это показывает, что гидродинамические свойства их макромолекул отличаются от соответствующих свойств гибких полимеров и требуют поэтому специального рассмотрения. [32]
Этот разброс объясняется различной степенью раз-ветвленности полученных гомополимеров. Среднее значение k составляет 0 43, что соответствует обычным значениям, получаемым для растворов гибких полимеров в термодинамически хороших растворителях. [33]
Увеличение молекулярной массы приводит к росту вязкости системы, что равносильно росту лсреохлаждепня, поэтому скорость зародышеобразовання повышается при снижении скорости роста кристаллов. Зависимость скорости кристаллизации от молекулярной массы имеет экстремальный характер Дтя высокорегулярных гибких полимеров увеличение молекулярной массы обычно ускоряет кристаллизацию, поско 1Ьку определяющим является снижение энергии зародышеобразовання за счет больше. Для менее гибких полимеров рост молекулярной массы может оказать негативное влияние на общую скорость кристаллизации из за снижения скорости роста кристалла вследствие высокой вязкости полимера. [34]
В основу теоретических уравнений было положено представление о гибких целях, однако уравнения Флори и Хаггинса часто применяют к растворам полимеров лрбой степени гибкости. Очевидно, по мере уменьшения гибкости цепей различие между вычисленной по уравнениям. Для очень разбавленных растворов даже эластичных гибких полимеров теоретические уравнения неприменимы, так как цепи свернуты в клубки, что расчетом не предусмотрено. [35]
С за счет увеличения полярности часто нивелируется вследствие снижения плотности. Дли мало-полярных органических полимеров модули упругости довольно близки. С и гнстере-зисных потерь ( рис. 5 18) Поэтому неполярные гибкие полимеры характеризуются низкими потерями и теплообразованием при эксплуатации в неравновесных условиях многократных деформаций. В равновесных условиях потери и модуль Сж не зависят от полярности полимера, а определяются лишь числом химических связей. [37]
 |
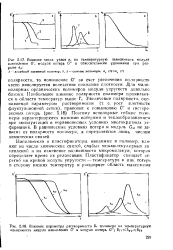
Температурная зависимость [ т ] и Л2 для системы с ВКТР ( а и. [38] |
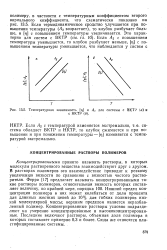
Концентрированными принято называть растворы, в которых молекулы растворенного вещества взаимодействуют друг с другом. В растворах полимеров это взаимодействие приводит к резкому увеличению вязкости по сравнению с вязкостью чистого растворителя. Нижний предел концентрации полимера в них может колебаться от доли процента для длинных жестких цепей до 10 % для гибких полимеров низкой молекулярной массы; верхним пределом является неразбавленный полимер. Концентрированные растворы условно подразделяют на умеренно концентрированные и высококонцентрированные. Последние включают растворы, объемная доля полимера в которых составляет примерно 0 3 и более. Сюда же относятся пластифицированные системы. [39]
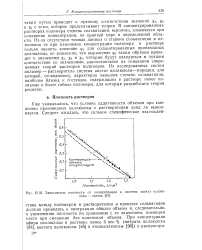 |
Зависимость плотности от концентрации в системе ацетат целлюлозы - ацетон. [40] |
В концентрированных растворах полимера степень сольватации, вероятно, изменяется при изменении концентрации, по крайней мере в определенной области. Из-за отсутствия точных данных о степени сольватации и изменении ее при изменении концентрации полимера в растворе нельзя оценить значение ф2 для сольватированных производных целлюлозы, но возможно, что выражение рг таким образом приведет к значениям хь Хл. Из исследованных систем полимер - растворитель система ацетат целлюлозы-пиридин, для которой, по-видимому, характерна меньшая степень сольватации, наиболее близка к системам, содержащим в растворе менее полярные и более гибкие полимеры, для которых разработана теория решетки. [41]
В очень интересном патенте [31] описывается полимеризация алифатических конъюгированных диолефинов на окислах металлов VIA группы, промотированных гидридами щелочноземельных металлов, например полимеризация бутадиена или изопрена на окисномолибденовом катализаторе в присутствии гидрида кальция. Согласно описаниям, образующийся полибутадиен содержит 20 % 1 2-звеньев, и 80 % 1 4-звеньев. Из числа последних 62 5 % имеют ис-конфигурацию и 37 5 % - / прайс-конфигурацию. Сополимеризация бутадиена и этилена дает прочные гибкие полимеры, по-видимому подобные полиэтилену. Однако, по данным инфракрасной спектроскопии, в полимере присутствуют - двойные связи, источником которых является бутадиен. Сополимер бутадиена и стирола, приготовленный тем же способом, что и полибутадиен, содержал 15 вес. [42]
В очень интересном патенте [31] описывается полимеризация алифатических конъюгированных диолефинов на окислах металлов VIA группы, промотированных гидридами щелочноземельных металлов, например полимеризация бутадиена или изопрена на окисномолибденовом катализаторе в присутствии гидрида кальция. Согласно описаниям, образующийся полибутадиен содержит 20 % 1 2-звеньев, и 80 % 1 4-звеньев. Из числа последних 62 5 % имеют ifttc - конфигурацию и 37 5 % - трансконфигурацию. Сополимеризация бутадиена и этилена дает прочные гибкие полимеры, по-видимому подобные полиэтилену. Однако, по данным инфракрасной спектроскопии, в полимере присутствуют двойные связи, источником которых является бутадиен. Сополимер бутадиена и стирола, приготовленный тем же способом, что и полибутадиен, содержал 15 вес. [43]
В работе О. Б. Птицына и Ю. А. Шаронова [6] было высказано предположение о том, что кристаллические конформации макромолекул в большинстве случаев определяются не межмолекулярными, а внутримолекулярными взаимодействиями. Это предположение основывалось первоначально на отмеченном выше ( см. гл. Бани и Холмз указали, что хотя кристаллическая структура макромолекул, вообще говоря, должна определяться не только внутри -, но и межмолекулярными взаимодействиями наиболее плотной упаковкой цепей [14]), однако максимальные различия между плотностями различных упаковок в углеводородных полимерах связаны с разностью энергий, равной всего - 300 кал / моль на звено. Поэтому межмолекулярное взаимодействие может определять кристаллическую структуру лишь достаточно гибких полимеров, в которых различные регулярные конформации отличаются на энергии, меньшие 300 кал / моль, а кристаллические структуры большинства полимеров должны определяться внутримолекулярными взаимодействиями. [44]
Флуоресцирующую молекулу можно ввести в аморфную область твердого кристаллического полимера или, если полимер находится в растворе, в растворитель. Тогда вращательное движение макромолекулы как целого или ее сегментов в случае гибких полимеров оказывает непосредственное влияние на флуоресцентное поведение системы. [45]
Страницы: 1 2 3 4