Полимеризация - тетрагидрофуран
Cтраница 3
 |
Зависимость In ( 1 - a / otp от In ( X / X0 ( М0 12 1 моль / л. Х0 0 1 моль / л. С0. [31] |
Таким образом, анализ молекулярных весов полимеров, полученных при полимеризации тетрагидрофурана в присутствии бутилацетата, показывает, что последний может взаимодействовать с растущей активной макромолекулой как агент передачи цепи и как сомономер. В пользу последнего вывода прямо свидетельствуют также данные кислотного гидролиза полимера. В соответствии с ожидаемым, молекулярный вес полимера после кислотного гидролиза в различных растворителях существенно уменьшается вследствие расщепления ортоэфирных связей, находящихся в сополимере. [32]
Это соединение, которое выделено в индивидуальном виде, инициирует полимеризацию тетрагидрофурана. [33]
 |
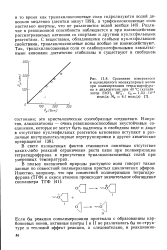
Кинетические кривые полимеризации тетрагидрофурана при 40 С под действием Et3O SbCl - ( C0 7 - 10 - 7 моль / л в присутствии добавок метилаля. [34] |
Следует отметить, что из-за экспериментальных трудностей не удается изучить полимеризацию тетрагидрофурана в присутствии достаточно больших добавок полидиоксолана, что не дает возможности более детально исследовать процесс передачи растущей цепи на молекулу полидиоксолана. [35]
Отметим, что доказательства отсутствия обрыва цепей были получены и при полимеризации тетрагидрофурана с другими каталитическими системами: галогенид металла а-окись [ Розенберг Б. А., Чеху т a, Людвиг Е. Б., Г а н т м а х е р, Медведев С. С., Высокомолек. Однако в последней работе автор указывает, что при высоких температурах расщепление комплексного проти-воиона, по-видимому, имеет место, что подтверждается наличием в полимере атомов фтора. [36]
На рис. 11.22 приведен типичный пример изменения вязкости реакционной смеси при полимеризации тетрагидрофурана. Как видно из рисунка, наблюдается сильное возрастание вязкости реакционной смеси после того, как основная масса мономера уже заполимери-зовалась. Расчеты показывают ( их принцип см. ниже), что столь значительное возрастание вязкости не может быть отнесено за счет увеличения длины полимерных цепей и концентрации полимера в реакционной массе. Отметим, что наблюдаемый эффект не обусловлен тиксотропными свойствами исследуемой системы, так как при следующих друг за другом измерениях происходило возрастание вязкости, а не падение, как это должно было бы быть, если бы эффект был вызван структурными изменениями системы. [37]
Показано [ 4, 11, 12, 161, что полимеризация тетрагидрофурана на карбок-сониевых активных центрах путем присоединения к атому кислорода отсутствует. [38]
Тем не менее, приведенные выше результаты определения характера изменения молекулярно-весового распределения при полимеризации тетрагидрофурана качественно подтверждают теоретический вывод, что обратимость роста живущих цепей практически не влияет на ширину молекулярно-весового распределения вплоть до глубоких стадий полимеризации. [39]
 |
Сравнение измеренного и вычисленного молекулярных весов при полимеризации тетрагидрофурана в дихлорэтане при 40 С ( катализатор ЕЮ BFT, С 1 53 - 10 - 2 моль / л. М 6 1 моль / л. [40] |
В свете изложенных фактов становится понятным отсутствие каких-либо реакций ограничения роста цепи при полимеризации тетрагидрофурана в присутствии триалкилоксониевых солей при умеренных температурах. [41]
Таким образом, изложенные выше факты однозначно свидетельствуют о том, что полиэфир при полимеризации тетрагидрофурана является агентом передачи цепи. [42]
Однако если в предыдущем случае не удалось выявить влияния добавок полидиоксолана на начальную скорость полимеризации тетрагидрофурана, то при добавлении метилаля ( 0 25 - 1 5 моль / л) это влияние очевидно ( рис. III.7), что может быть объяснено, с одной стороны, большей основностью метилаля, с другой, - более высокой концентрацией добавки. [43]
Другой метод полимеризации 1 4-эпокснннклогексана предусматривает использование сложного катачнчаторя ( хлорное железо - тионилхлорид), который, как было показано, эффективен также для полимеризации тетрагидрофурана. Так, к 50 г эпокснциклогексана, охлажденному до 0е, добавляют 0 015 г безводного хлорного железа в виде 10 % - ного растворл в эфире и 0 062 г тионилхлорида также в виде 10 % ного раствора в безводном эфире. Смесь перемешивают и выдерживают в закрытой колбе при 0 в течение 18 час. После этого полимер смешивают со спиртом и твердый продукт отфильтровывают от непрореагировавшего мономера. Полимер промывают повторно спиртом и водой и высушивают. [44]
 |
Влияние щелочного гидролиза. [45] |
Страницы: 1 2 3 4