Ван-дро
Cтраница 2
Работы Ван-дер - Ваальса и ван Лаара. Иной подход к теории растворов был развит голландским физиком Ван-дер - Ваальсом. Ван-дер - Ваальс попытался обобщить уравнение состояния, выведенное им для чистых жидкостей и газов, на случай растворов. Идеи Ван-дер - Ваальса получили дальнейшее развитие в работах ван Лаара. [16]
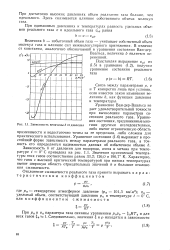 |
Зависимость величины b от давления. [17] |
Уравнение Ван-дер - Ваальса не дает удовлетворительной точности при вычислении параметров состояния реального газа. Уравнения состояния, предложенные многими другими исследователями, либо имеют ограниченную область применимости и недостаточно точны за ее пределами, либо сложны для практического использования. Зависимость b от давления для водорода, азота и метана при температуре t 0 С приведена на рис. 1.1. Значения критической температуры этих газов соответственно равны 33 2; 126 0 и 190 7 К. Характерно, что газы с высокой критической температурой при низких температурах имеют широкую область отрицательных значений b и большие отклонения сжимаемости. [18]
Силы Ван-дер - Ваальса представляют собой сумму этих сил притяжения. [19]
Открытия Ван-дер - Керка и Люитена в настоящее время используются в промышленном масштабе. [20]
Уравнение Ван-дер - Ваальса хорошо согласуется с опытом в тех случаях, когда пасует уравнение Клапейрона-Менделеева. [21]
Силы Ван-дер - Ваальса, обусловливающие физическую адсорбцию, включают дисперсионные, ориентационные и индукционные силы притяжения молекул к твердой поверхности. В зависимости от структуры молекул и свойств твердого тела преобладает та или иная сила притяжения. В случае металлического кристалла большое влияние на адсорбцию оказывают электростатические силы притяжения. [22]
Уравнение Ван-дер - Ваальса в безразмерных координата) позволяет вычислить коэффициенты разложения свободной энергии (1.22) в теории Ландау. [23]
Силы Ван-дер - Ваальса часто заметно проявляются во взаимодействии атомов, молекул, малых частичек конденсированной фазы, макроскопических конденсированных тел. Эти силы обычно доминируют на достаточно больших расстояниях, когда перекрытие волновых функций взаимодействующих тел не слишком существенно. По сравнению с характерными энергиями химической связи энергия ван-дер-ваальсова взаимодействия мала. Поэтому, в частности, для его детального экспериментального изучения требуется довольно высокая точность измерений. [24]
Уравнение Ван-дер - Ваальса ( с фиксированными значениями а и 6) в этом случае непригодно, и при вычислении интеграла необходимо пользоваться экспериментальными данными о поведении параметров газа во всем диапазоне их изменения в процессе дросселирования. [25]
Модель Ван-дер - Ваальса, в которой дальнодействующие силы учитываются, верно описывает достаточно плотный газ. Естественно распространить эту модель в область еще более высоких плотностей, когда газ переходит в жидкость. [26]
Силы Ван-дер - Ваальса можно вычислить, если рассмотреть взаимодействие достаточно удаленных друг от друга атомов. На большом расстоянии между атомами валентные силы, вычисляемые из первого приближения теории возмущения, очень малы. Напротив, на этих расстояниях оказывается уже невозможным игнорировать второе приближение, в котором учитывается деформация электронных оболочек атомов. [27]
Уравнение Ван-дер - Ваальса с большой точностью описывает свойства разреженных газов при достаточно высоких температурах. Но в плотном газе и при температурах, близких к температуре конденсации, это уравнение в количественном отношении неудовлетворительно. Поэтому неоднократно предлагались другие, более точные уравнения состояния реальных газов. Широкое использование уравнения Ван-дер - Ваальса связано, во-первых, с его относительно простой математической структурой, а во-вторых, с тем, что оно качественно правильно передает свойства плотных газов и содержит указания на переход в жидкое состояние и критические явления. [28]
Изотермы Ван-дер - Ваальса на pF - диаграмме с повышением температуры поднимаются вверх. Это сопровождается сближением объемов YI и F2 определяющих соответственно объемы жидкой и газообразной фазы. Совпадение жидкой и газообразной фазы произойдет в точке А ( см. рис. 10), которая называется критической. Эта точка определяется точкой перегиба изотермы, температура которой называется критической. [29]
Уравнение Ван-дер - Ваальса и все его последующие модификации являются кубическими относительно объема, поэтому в результате его решения получают один или три действительных корня. При рассмотрении паровой фазы ее объем соответствует максимальному положительному действительному корню, а при рассмотрении жидкой фазы ее объему соответствует минимальный положительный действительный корень уравнения состояния. [30]
Страницы: 1 2 3 4