Величина - энергия - активация - реакция
Cтраница 3
При покрытии никелевого катализатора кислородом ( играющим в этих условиях роль необратимого яда) в количестве до 15 - 20 % от монослойного величины энергии активации реакций пара-орто-конверсии водорода и изотопного обмена водорода с дейтерием при комнатной температуре повышались, а при дальнейшем покрытии изменялись практически мало. Этот эффект был объяснен проникновением кислорода, покрывающего места средней и малой активности, в глубь поверхности, с образованием более прочного его соединения. [31]
Большим успехом радикально-цепной теории крекинга алка-нов явилось объяснение первого порядка валовой реакции распада и вычисление эффективной энергии активации валового процесса крекинга на основании разумных предположен ний о величинах энергий активации реакций зарождения, развития и обрыва цепей. В случае отдельных алканов эти оценки приводили к хорошему согласию с опытно определяемой валовой энергией активации крекинга. Измеренные концентрации радикалов значительно превышали равновесные концентрации их, которые соответствуют диссоциационным равновесиям в реакциях Н2 2Н или СН4 СН3 Н при температурах крекинга, что является прямым доказательством участия радикалов в процессе. [32]
Приведенный анализ позволяет сделать вывод, что на поверхности серебра может присутствовать значительное количество деформаций, не оказывающих никакого влияния на ее каталитическую активность; увеличение деформации должно приводить к возрастанию значений А и Е только при условии, что деформация дает большое число центров, величины энергии активации реакции на которых близки к значению Ег. Используя данные табл. 5.1, можно выразить наши выводы в количественной форме. При напряжении 22 5 в для грани ( 111) с2 0, как и раньше, ci 106 центров на 1 см2, причем для реакции на этих центрах энергия активации равна 12 ккал-молъ-1. Изменения величины А в 105 раз и Е на 10 кпал-молъ 1 после бомбардировки ионами при напряжении, равном 4000 в, можно теперь объяснить правдоподобным предположением, что на 1 см2 образовалось 10е - 105 10й центров; причем для реакции на каждом центре энергия активации составляла. Следовательно, если первоначально имевшиеся на 1 см2 10е центров после интенсивной бомбардировки остаются на грани ( 111), то их влияние должно маскироваться присутствием 1011 новых дислокаций, образовавшихся на каждом квадратном сантиметре поверхности. [33]
Уже в прежних работах по анионной полимеризации стирола и других мономеров была установлена возможность практически полного подавления диссоциации ионных пар на свободные ноны путем введения в систему буферных соединений, в частности тет-рафенилборнат. Величины энергии активации реакции роста выше и ниже этой температуры составляют соответственно 2 и 7 ккал / моль. Эти данные приводят к выводу об изменении типа ионных пар с температурой. В еще более явном виде переход одного типа ионных тар в другой проявляется при полимеризации в малополярной среде в присутствии небольших количеств сильных оснований Льюиса. [34]
В работе [634] показано, что при разложении циклогексана на никелевых и платиновых катализаторах, нанесенных на окись алюминия, уменьшение, электропроводности катализаторов соответствует увеличению выхода ароматических углеводородов. При этом величины энергии активации реакции и энергии активации электропроводности оказываются близкими, что, по мнению авторов [634], свидетельствует о связи каталитических и электронных свойств. [35]
Низкое значение величины энергии активации реакции полимеризации в сравнении с энергиями активации реакции-разложения парафинов ( 60 600 - 65 000 кал) объясняет преобладание реакций полимеризации при умеренной температурной обработке углеводородных газов, содержащих парафины и олефины. [36]
Рогинский учитывает взаимосвязь величин энергии активации реакции, изменения работы выхода электрона и степени покрытия поверхности катализатора. Так, если скорость процесса определяется скоростью адсорбции одно-го из веществ, то из уравнений (III.241), (III.242), (III.250) и (III.256) получаются уравнения кинетики с дробными степенями, обусловленными изменением работы выхода электрона в результате адсорбции. [37]
Сравнение величин константы скорости термического разложения показывает, что как в случае диацилперекисей, так и в случае перэфиров скорость термического разложения резко возрастает при увеличении степени замещения углеродного атома, непосредственно связанного с карбоксигруппой. Соответственно наблюдается понижение величины энергии активации реакции термического разложения. [38]
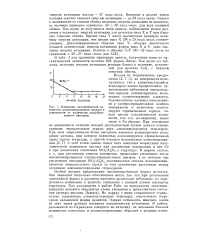 |
Изменение каталитической активности декатионировапного цеолита в зависимости от количества адсорбированного хинолина. [39] |
В табл. 3 для сравнения приведены данные, полученные нами по каталитической активности цеолита 10Х фирмы Линде. Как видно из таблицы, величина энергии активации реакции близка к величине, полученной для цеолита СаХ, с близкой степенью обмена. [40]
При механическом воздействии на высокомолекулярное вещество энергия расходуется на деформацию валентных углов и на разрыв полимерной цепи. Эти процессы вызывают уменьшение - величины энергии активации реакций химического превращения полимеров, подвергающихся действию сил агрессивной среды. При действии механических аил деструкция протекает по радикальному или ионному механизму, или же оба механизма могут сочетаться. [41]
При этом зависимость величины lg ф ( характеристики размеров поджигающего тела и скорости газа) от l / Ts должна быть линейной. Угловой коэффициент этой прямой определяет величину энергии активации реакции в пламени. Было установлено, что в некоторых случаях эти выводы теории выполняются, и зависимость lg ф от ITS дает разумное значение А. Однако чаще эта зависимость оказывается нелинейной, вычисляемая величина А неправдоподобна, а пределы поджигания, вопреки утверждениям теории, зависят от материала поджигающего тела. [42]
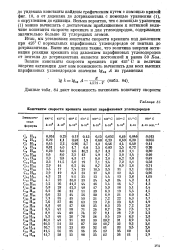 |
Константы скорости крекинга высших парафиновых углеводородов. [43] |
Итак, мы установили константы скорости крекинга под давлением при 425 С нормальных парафиновых углеводородов от пентана до дотриаконтана. Выше мы приняли также, что величина энергии активации реакции крекинга под давлением парафиновых углеводородов от пентана до дотриаконтана является постоянной и равна 69 Кал. [44]
IB частности, перекисью лаурила [242, 243], приводит к восстановлению хиноидных групп, исчезновению индукционного периода полимеризации и уменьшению величины энергии активации реакции инициирования. В этих условиях, кроме привитой полимеризации на поверхности сажи, происходит и гомополимеризация, инициируемая перешедшими в раствор мономера радикалами. Аналогичные результаты получены в работе [244], в которой также показано, что в случае диа-рильных перекисей ингибирующее влияние сажи на полимеризацию Ненасыщенных полиэфиров выражено сильнее. [45]
Страницы: 1 2 3 4