Расчет - электронная структура
Cтраница 2
Из расчета электронной структуры молекулы диметилсульфоксида, проведенного методом ССП МО ЛКАО в приближении полного пренебрежения дифференциальным перекрыванием, вытекает [350], что р - - орбитали кислорода обладают ярко выраженной донорной способностью. [16]
Результаты расчетов электронной структуры комплексов платины типа [ PtLX3 ] n позволили также более конкретно подойти к вопросу о соотношении о-донорных и л-акцепторных свойств лигандов. Для лигандов же типа CN -, CO была обнаружена антипараллельность хода кривых Va и Ул при варьировании состава комплекса. Объяснение этого кажущегося расхождения с обычной теорией кроется в том, что последняя фактически рассматривает изолированную систему металл-лиганд L и не учитывает присутствия остальных лигандов. Действительно, если лиганды X являются слабыми а-донорами ( X - Н20), то 0-донорная активность лп-ганда L будет проявляться без всяких помех. Если же лиганды X - сильные доноры, то их присутствие будет стимулировать перенос электронной плотности с металла на пустые л-орбитали лиганда L. Это, в свою очередь, вызовет усиление о-донорной способности рассматриваемого лиганда, но их проявление будет затруднено вследствие наличия мощного встречного потока заряда с лигандов X. Реально могут встретиться и более сложные случаи, особенно в комплексах смешанного состава, так что, вообще говоря, соотношение между донорными и акцепторными свойствами одного и того же лиганда может быть разным в каждом конкретном случае. [17]
Для расчета электронной структуры сложных молекул метод МО ЛКАО в наиболее общей форме был развит Рутаном [75, 85, 86] на основе идей Хартри и Фока. Отличие состоит в том, что матричные элементы Hv включают наряду с молекулярными интегралами типа (4.5) и (4.6), которые могут быть вычислены, коэффициенты Сц1, которые неизвестны с самого начала. Я ц и е, а затем по е с помощью (4.3) отыскивается новый набор c vi, и такая процедура повторяется до совпадения предыдущего результата с последующим. [18]
При расчете электронной структуры кристалла в модели КРЭЯ на основе приближения ЛКАО исходными являются уравнения (3.30), которые решаются теперь в соответствии со сказанным выше только при нулевом значении вектора k, однако ап в блоховских суммах нумерует теперь расширенные элементарные ячейки. Поэтому изменяется и нумерация базисных функций Фр ( г) - число их теперь определяется числом атомных функций не в минимальной, а в расширенной ячейке. [19]
Проведенный предварительно расчет электронной структуры молекулы НСООН показал, что конформация с протонами в транс-положении ( син-конформация) имеет энергию ниже на 20 5 кДж / моль, чем анти-онформация с протонами в цшмюяожении. [20]
На основе первопринципных расчетов электронной структуры изучена относительная устойчивость Ьсс, fee, hep, CslV и Cmca кристаллических структур натрия под давлением при Г0 К. Учтена делокализация 2s - и 2р - электронов натрия при сжатии и изменение под давлением параметров, определяющих базис решеток Бравэ рассмотренных кристаллических структур. [21]
Приближенные методы расчета электронной структуры твердых тел на базисе ЛКАО, возможности которых выяснены на сравнительно простых системах, оказываются весьма полезными при рассмотрении более сложных объектов: ламеляр-ных ( слоистых) соединений, хемосорбции атомов и молекул на поверхностях твердых тел и др. Особенно перспективны они в теории кристаллов с глубокими локальными центрами - это показывают все полученные для них результаты, обсуждаемые в пятой главе. [22]
На основании расчетов электронной структуры гидридных комплексов Pd ( II), Pi ( II) и N1 ( 11), проведено сравнение способности этих соединений к реакциям диссоциации и замещения о образованием гидрид-иона. Обсуждены возможные причины большей эф ектив-ности в каталитических реакциях гидрирования комплексов палладия и платины яо сравнению с комплексами никеля. [23]
Таким образом, расчет электронной структуры относительно небольшой КРЭЯ B3N3 уже позволяет оценить з самосогласованном расчете наиболее важные состояния совершенного кристалла BNreHC - границы энергетических зон. [24]
Использование ЭВМ для расчета электронной структуры молекул позволяет получить важные качественные и количественные сведения относительно простейших многоэлектронных молекул. [25]
 |
Электронные характеристики, полученные методом ЛКАО МО и связанные с ними свойства. [26] |
Методами квантовой химии проведены расчеты электронных структур важнейших классов биологических молекул. [27]
Однако последующие спектральные исследования и расчеты электронной структуры показали, что эти представления не соответствуют действительности, так как распределение электронной плотности по хелатному циклу неравномерно. [28]
Цумдал и Драго на основании расчетов электронной структуры комплекса m / 7aHc - [ Pt ( PH3) ( NH3) Q2 ] пришли к заключению, что я-составляющая в связи Pt - Р равна всего лишь 15 % от ст-составляю-щей. Следует заметить, что для расчета они использовали модификацию расширенного метода Хюккеля, предложенную Хоффманом, в которой, как и в любом полуэмпирическом методе, получаемые результаты в большой степени зависят от выбора используемых параметров. [29]
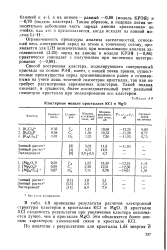 |
Кластерные модели кристаллов КС. и MgO. [30] |
Страницы: 1 2 3 4 5