Молекулярный расчет
Cтраница 4
 |
Результаты расчета NiFe-4 в приближении ППДП по данным. [46] |
Все эти подробности еще раз подчеркивают несовершенство полуэмпирических подходов при проведении молекулярных расчетов. [47]
Существует ряд обзоров, посвященных базисным наборам, которые удобно использовать в разнообразных применениях. Чарский и Урбан [86] описали использование слейтеровских и гауссовых базисных наборов в молекулярных расчетах, посвященных установлению различных свойств. [48]
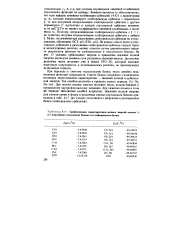 |
Сравнительные характеристики полных энергий атомов Li и F в различных гауссовских базисах и в слейтеровском базисе. [49] |
На вопрос, какое число р-гаус-совских орбиталей в разложении слейтеровской орбитали следует брать в молекулярных расчетах, можно ответить путем сравнительного анализа результатов расчетов на слейтеровском и гауссовском базисах. Достаточно разумные числа получают уже в базисе STO - 3G, который завоевал известную популярность в неэмпирических расчетах, не претендующих на большую точность. [50]
В реальных расчетах и точная волновая функция в рамках выбранной пробной модели, и сама точная волновая функция остаются неизвестными. При вычислении ожидаемого значения гамильтониана для пробной волновой функции принято ( по крайней мере в молекулярных расчетах) параметризовать орбитали при помощи конечного базисного набора. Это неизбежно приводит к ошибке, связанной с эффектами усечения базисного набора ( см. гл. При вычислении корреляционной поправки к волновой функции Ф, приходится не только использовать конечный базисный набор, но также ограничиваться лишь частью членов, необходимых для полного описания эффектов электронной корреляции в рамках данного базисного набора. На рис. 3.3 схематически изображено соотношение между результатами реальных расчетов, точными результатами пробной модели и точными результатами с учетом корреляционной поправки. [51]
Но здесь следует иметь в виду одно важное обстоятельство: те АО, которые используются в молекулярных расчетах, - это не орбитали, полученные причисленном интегрировании уравнений Хартри - Фока для атомов. Последние заданы не в аналитическом, а в табличном виде, и поэтому крайне неудобны для молекулярных расчетов. Это значит, что необходимо каким-то образом аппроксимировать АО. [52]
В настоящее время в квантовой химии используют главным образом два типа базисных функций, каждый из которых отдает предпочтение одному из двух указанных выше критериев. Функции обоих типов являются одноцентровыми, т.е. они существенно привязаны к определенной точке пространства - центру и обладают некоторой симметрией относительно своего центра. В молекулярных расчетах почти всегда используют наборы базисных функций, центрированных на разных точках пространства. Чаще всего в качестве центров берутся атомные ядра, хотя базисную функцию можно центрировать и на связи. В некоторых случаях используют функции с плавающим центром, т.е. точки центрирования базисных функций не фиксируют заранее, а подбирают в процессе расчета так, чтобы получить оптимальные результаты. [53]
Но здесь следует иметь в виду одно важное обстоятельство: те АО, которые используются в молекулярных расчетах, - это не орбитали, полученные причисленном интегрировании уравнений Хартри - Фока для атомов. Последние заданы не в аналитическом, а в табличном виде, и поэтому крайне неудобны для молекулярных расчетов. Это значит, что необходимо каким-то образом аппроксимировать АО. [54]
Приложение I напоминает читателю те основные положения квантовой механики, которые используются в основном тексте книги. В приложении II собраны формулы для функций атомных орбиталей, с которыми постоянно приходится сталкиваться каждому, кто работает в области молекулярных расчетов. Приложение III-это пояснение основных положений теории групп в той мере, насколько знание их необходимо для понимания основного текста книги. Обычно такие взаимодействия не рассматривают в учебных руководствах, хотя они приобретают все большее значение в связи с успехами экспериментов по электронному и ядерному резонансу. [55]
Опыт большого числа расчетов электронного строения молекул показывает, что вычисление молекулярных интегралов занимает большую часть времени, необходимого для решения задачи. В связи с этим особую важность приобретает разработка эффективных методов быстрого и точного расчета требуемых интегралов. В настоящей работе описаны способы вычисления одно -, двух - и трехцентровых одноэлектронных и одно - и двухцентровнх ку-лоновских интегралов, используемых в неэмпирических молекулярных расчетах. [56]
Страницы: 1 2 3 4